Informace Na Stránce Nepředstavují Lékařskou Radu. Nic Neprodáváme. Přesnost Překladu Není Zaručena. Zřeknutí Se Odpovědnosti
Antipsariatics, SystemicHumira
Shrnutí drog
Co je to Humira?
Humira (adalimumab) je injekční protein (protilátka) používaný k léčbě revmatoidní artritidy juvenilní idiopatic artritida Psoriatická artritida Ankylozující spondylitida a Plakční psoriáza. Humira se také používá k léčbě Crohnovy choroby poté, co byly ostatní léky vyzkoušeny bez úspěšné léčby symptomů.
Jaké jsou vedlejší účinky pro Humiru?
Mezi běžné vedlejší účinky humiry patří
- Reakce místa injekce (zarudnutí svědění bolesti modřin nebo krvácení)
- bolest hlavy
- Suppy nos
- sinusová bolest nebo
- bolest žaludku.
Řekněte svému lékaři, pokud máte vážné vedlejší účinky Humira, včetně:
- Rychlý/nepravidelný/bušící srdeční rytmus
- bolest žaludku
- krev ve stolici
- Změny mentální/nálady
- Těžká bolest hlavy
- Snadné modřiny nebo krvácení
- Tmavá moč
- žloutnutí očí a kůže
- Bolest nohou nebo otok
- otupělost nebo brnění paží/rukou/nohou/nohou
- Nestavitost
- nevysvětlitelná svalová slabost
- potíže s mluvením/žvýkání/polykání/pohyby obličeje
- změny vize
- Extrémní únava
- bolest kloubů nebo
- Vyrážka ve tvaru motýla na nose a tvářích.
Dávkování pro Humiru
Doporučená dávka Humiry u dospělých pacientů s revmatoidní artritidou (RA) psoriatickou artritidou (PSA) nebo ankylozující spondylitidou (AS) je 40 mg podávána každý druhý týden. Pediatrická dávka je určována hmotností dítěte.
Jaké léky nebo doplňky interagují s Humirou?
Jiné léky mohou interagovat s Humirou. Sdělte svému lékaři veškeré léky a doplňky, které používáte, na předpis a volně prodejné léky.
Humira During Těhotenství a Breastfeeding
Během těhotenství by měla být Humira použita pouze tehdy, když je předepsána. Není známo, zda tento lék prochází do mateřského mléka. Podobné drogy procházejí do mateřského mléka. Před kojením se poraďte se svým lékařem.
Další informace
Naše Hutira (Adalimumab) vedlejší účinky Drug Center poskytuje komplexní pohled na dostupné informace o drogách o možných vedlejších účincích při užívání tohoto léku.
Informace o drogách FDA
- Popis léku
- Indikace
- Dávkování
- Vedlejší účinky
- Lékové interakce
- Varování
- Předávkovat
- Klinická farmakologie
- Průvodce léky
VAROVÁNÍ
Vážné infekce a malignita
Vážné infekce
Pacienti léčeni Humirou jsou vystaveni zvýšenému riziku rozvoje závažných infekcí, které mohou vést k hospitalizaci nebo smrti [viz varování a OPATŘENÍ ]. Většina pacientů, kteří se tyto infekce vyvinuli, užívala doprovodné imunosupresivy, jako je methotrexát nebo kortikosteroidy.
Přerušte humiru, pokud se pacient vyvine vážnou infekci nebo sepse.
Hlášené infekce zahrnují:
- Aktivní tuberkulóza (TB) včetně reaktivace latentní TB. Pacienti s TBC často představovali diseminované nebo extrapulmonální onemocnění. Otestujte pacienty na latentní TB před použitím humiry a během terapie. Před použitím humira zahájíte léčbu latentní TB.
- Invazivní plísňové infekce včetně histoplazmózy kokcidioidomykózy kandidóza aspergilóza blastomykóza a pneumocystosis. Pacienti s histoplazmózou nebo jinými invazivními plísňovými infekcemi mohou vyskytovat spíše diseminované než lokalizované onemocnění. U některých pacientů s aktivní infekcí může být testování antigenu a protilátek na histoplazmózu negativní. Zvažte empirickou terapii proti omylu u pacientů s rizikem invazivních plísňových infekcí, kteří se rozvíjejí závažné systémové onemocnění.
- Bakteriální virové a další infekce způsobené oportunními patogeny včetně Legionella a Listeria.
Pečlivě zvažte rizika a přínosy léčby Humirou před zahájením terapie u pacientů s chronickou nebo opakující se infekcí.
Pečlivě sledujte pacienty pro rozvoj příznaků a příznaků infekce během a po léčbě humirou, včetně možného vývoje TB u pacientů, kteří testovali negativní na latentní infekci TB před zahájením terapie [viz varování a varování a OPATŘENÍ a Nežádoucí účinky ].
Malignita
Lymfom a další malignity Některé fatální byly hlášeny u dětí a dospívajících pacientů léčených blokátory TNF včetně humira [viz varování a OPATŘENÍ ]. Post-marketing cases of hepatosplenic T-cell lymphoma (HSTCL) a rare type of T-cell lymphoma have been reported in patients treated with TNF blockers including Humira. These cases have had a very aggressive disease course a have been fatal. The majority of reported TNF blocker cases have occurčervený in patients with Crohn's disease or ulcerative colitis a the majority were in adolescent a young adult males. Almost all these patients had received treatment with azathioprine or 6-mercaptopurine (6–MP) concomitantly with a TNF blocker at or prior to diagnosis. It is uncertain whether the occurrence of HSTCL is related to use of a TNF blocker or a TNF blocker in combination with these other immunosuppressants [see VAROVÁNÍS AND OPATŘENÍ ].
Popis pro Humiru
Humira (adalimumab) je rekombinantní lidská monoklonální protilátka IgG1 specifická pro faktor lidské nádorové nekrózy (TNF). Humira byla vytvořena pomocí technologie displeje ve fázi, která vedla k protilátce s lidskými těžkými a lehkými řetězovými proměnlivými oblastmi a lidskými IgG1: K konstantními oblastmi. Adalimumab je produkován technologií rekombinantní DNA v systému exprese savčích buněk a je purifikován procesem, který zahrnuje specifické virové inaktivace a odstranění kroků. Skládá se z 1330 aminokyselin a má molekulovou hmotnost přibližně 148 kilodaltonů.
Humira je dodávána jako sterilní řešení bez konzervační látky adalimumabu pro podkožní podání. Produkční produkt je dodáván buď jako předběžné pero s jedním použitím (HUMIRA) jako 1 ML Skleněné injekční injekční injekční institucionální institucionální použití 1 ml. Unclosed uvnitř pera je jedno použití 1 ml předplacené skleněné stříkačky. Roztok humiry je jasný a bezbarvý s pH asi 5,2.
Každá předběžná stříkačka nebo předplněná pero 80 mg/0,8 ml dodává 0,8 ml (80 mg) léčivého přípravku. Každý 0,8 ml Humira obsahuje polysorbát 80 (NULL,8 mg) a vodu pro injekci USP adalimumab (80 mg) mannitol (NULL,6 mg).
Každý 40 mg/0,4 ml předplněné stříkačky nebo předplněné pero poskytuje 0,4 ml (40 mg) léčivého přípravku. Každý 0,4 ml Humira obsahuje polysorbát 80 (NULL,4 mg) a vodu pro injekci USP adalimumab (40 mg) mannitol (NULL,8 mg).
Každé 40 mg/0,8 ml předplněné stříkačky předplněné pero nebo jedno použití institucionální používání poskytuje 0,8 ml (40 mg) léčivého produktu. Každý 0,8 ml Humira obsahuje monohydrát kyseliny citronové adalimumab (40 mg) (NULL,04 mg) dibasic sodný fosfát dihydrát (NULL,22 mg) Mannitol (NULL,6 mg) Monobasický fosfátový fosfát fosfát (NULL,93 mg) sodný (NULL,93 mg) sodný (NULL,93 mg) sodík) sodík) sodík) sodík) sodík) sodík) (NULL,8 mg) sodík). Citrát sodný (NULL,24 mg) a voda pro injekční USP. Pro úpravu pH se přidává hydroxid sodný.
Každá předběžná stříkačka 20 mg/0,2 ml dodává 0,2 ml (20 mg) léčivého produktu. Každý 0,2 ml Humira obsahuje polysorbát 80 (NULL,2 mg) a vodu pro injekci USP adalimumab (20 mg) mannitol (NULL,4 mg).
Každá předběžná stříkačka 20 mg/0,4 ml dodává 0,4 ml (20 mg) léčivého produktu. Každý 0,4 ml Humira obsahuje monohydrát kyseliny citronové adalimumab (20 mg) (NULL,52 mg) dibasic sodný fosfát dihydrát (NULL,61 mg) mannitol (NULL,8 mg) monobasický fosfát fosfát (NULL,8 mg) sodík (NULL,8 mg) sodík (NULL,8 mg) sodík) sodík (NULL,8 mg) sodík) sodný) sodný (NULL,8 mg) sodík (NULL,8 mg) sodík) sodný (NULL,8 mg) sodík (NULL,8 mg) sodík (NULL,8 mg) sodík) sodný (NULL,8 mg) sodík) (NULL,8 mg) (NULL,8 mg) (NULL,8 mg) (NULL,8 mg) (NULL,8 mg) (NULL,8 mg) (NULL,8 mg) ( 2,8 mg) (NULL,8 mg) (NULL,8 mg). Citrát (NULL,12 mg) a voda pro injekční USP. Pro úpravu pH se přidává hydroxid sodný.
Každých 10 mg/0,1 ml předplněné stříkačky dodává 0,1 ml (10 mg) léčivého produktu. Každý 0,1 ml Humira obsahuje polysorbát 80 (NULL,1 mg) a vodu pro injekci USP.
Každých 10 mg/0,2 ml předplněné stříkačky dodává 0,2 ml (10 mg) léčivého produktu. Každý 0,2 ml Humira obsahuje monohydrát kyseliny citronové adalimumab (10 mg) (NULL,26 mg) dibasic sodný fosfát dihydrát (NULL,31 mg) mannitol (NULL,4 mg) monobasický fosfát fosfát (NULL,23 mg) sodný (NULL,2 mg) sodný (NULL,2 mg) sodný (NULL,2 mg) sodný) sodný) sodný) sodík) sodík) (NULL,4 mg) sodík) sodík (NULL,4 mg) sodík (NULL,2 mg) sodík (NULL,2 mg) sodík (NULL,2 mg). Citrát (NULL,06 mg) a voda pro injekční USP. Pro úpravu pH se přidává hydroxid sodný.
Použití pro Humiru
Revmatoidní artritida
Humira je indikována pro snižování příznaků a symptomů vyvolávajících hlavní klinickou odpověď inhibující progresi strukturálního poškození a zlepšení fyzické funkce u dospělých pacientů s mírně až vážně aktivní revmatoidní artritidou. Humira lze použít samostatně nebo v kombinaci s methotrexátem nebo jinými nebiologickými anti-rheumatickými léky modifikujícími onemocnění (DMARD).
Juvenilní idiopatická artritida
Humira je indikována pro snížení příznaků a symptomů mírně na těžce aktivní polyartikulární juvenilní idiopatickou artritidu u pacientů ve věku 2 let a starší. Humira lze použít samostatně nebo v kombinaci s methotrexátem.
Psoriatická artritida
Humira je indikována pro snižování příznaků a symptomů inhibující progresi strukturálního poškození a zlepšení fyzické funkce u dospělých pacientů s aktivní psoriatickou artritidou. Humira lze použít samostatně nebo v kombinaci s nebiologickými DMARDS.
Ankylozující spondylitida
Humira je indikována pro snižování příznaků a symptomů u dospělých pacientů s aktivní ankylozující spondylitidou.
Humira je indikována pro léčbu mírně až vážně aktivní Crohnovy choroby u dospělých a dětských pacientů ve věku 6 let a starších.
Ulcerativní kolitida
Humira je indikována pro léčbu mírně až vážně aktivní ulcerativní kolitidy u dospělých a dětských pacientů ve věku 5 let a starších.
Omezení použití
Účinnost Humiry nebyla stanovena u pacientů, kteří ztratili odpověď nebo byli netolerantní na blokátory TNF [viz Klinické studie ].
Plakesová psoriáza
Humira je indikována pro léčbu dospělých pacientů se středně těžkou až těžkou chronickou plakovou psoriázou, kteří jsou kandidáty na systémovou terapii nebo fototerapii a kdy jsou jiné systémové terapie lékařsky méně vhodné. Humira by měla být podávána pouze pacientům, kteří budou pečlivě sledováni a budou mít pravidelné následné návštěvy u lékaře [viz VAROVÁNÍS AND OPATŘENÍ ].
Hidradenitis Supsurative
Humira je indikována pro léčbu střední až těžké hidradenitidy Supsurativa u pacientů ve věku 12 let a starších.
Uveitida
Humira je indikována pro léčbu neinfekčních meziproduktů a panuveitidy u dospělých a pediatrických pacientů ve věku 2 let a starších.
Dávkování pro Humiru
Revmatoidní artritida Psoriatická artritida And Ankylozující spondylitida
Doporučená subkutánní dávka Humiry u dospělých pacientů s revmatoidní artritidou (RA) psoriatickou artritidou (PSA) nebo ankylozující spondylitidou (AS) je podávána 40 mg každý druhý týden. Během léčby Humirou může pokračovat další nebiologické glukokortikoidy (MTX) Jiné nebiologické glukokortikoidy Glukokortikoidy Nesteroidální protizánětlivé léky (NSAID) a/nebo analgetiky. Při léčbě RA mohou někteří pacienti, kteří neužívají souběžný MTX, získá další prospěch ze zvýšení dávky humiry na 40 mg každý týden nebo 80 mg každý druhý týden.
Juvenilní idiopatická artritida Or Dětská uveitida
Doporučená subkutánní dávka Humira pro pacienty ve věku 2 let a starší s polyartikulární juvenilní idiopatickou artritidou (JIA) nebo pediatrickou uveitidou je založena na hmotnosti, jak je ukázáno níže. MTX glukokortikoidy NSAID a/nebo analgetika mohou pokračovat během léčby Humirou.
| Pediatrická hmotnost (2 roky a starší) | Doporučené dávkování |
| 10 kg (22 liber) na méně než 15 kg (33 liber) | 10 mg každý druhý týden |
| 15 kg (33 liber) na méně než 30 kg (66 liber) | 20 mg každý druhý týden |
| 30 kg (66 liber) a větší | 40 mg každý druhý týden |
Humira nebyla studována u pacientů s polyartikulární JIA nebo pediatrickou uveitidou mladší než 2 roky nebo u pacientů s hmotností pod 10 kg.
Dospělí
Doporučená subkutánní dávka Humiry pro dospělé pacienty s Crohnovou chorobou (CD) je 160 mg zpočátku v den 1 (poskytnuto za jeden den nebo rozděleno po dobu dvou po sobě následujících dnů) následuje 80 mg o dva týdny později (15). O dva týdny později (29. den) začínají dávkování 40 mg každý druhý týden. Aminosalicyláty a/nebo kortikosteroidy mohou pokračovat během léčby Humirou. Azathioprin 6-merkaptopurin (6-MP) [viz VAROVÁNÍS AND OPATŘENÍ ] nebo MTX může pokračovat během léčby Humirou v případě potřeby.
Pediatrie
Doporučená subkutánní dávka Humira u dětských pacientů ve věku 6 let a starších s Crohnovou chorobou (CD) je založena na tělesné hmotnosti, jak je ukázáno níže:
| Pediatrická váha | Doporučené dávkování | |
| Dny až 15 | Počínaje 29. den | |
| 17 kg (37 liber) na méně než 40 kg (88 liber) | Den 1: 80 mg den 15: 40 mg | 20 mg každý druhý týden |
| 40 kg (88 liber) a větší | Den 1: 160 mg (jediná dávka nebo rozdělení po dobu dvou po sobě jdoucích dnů) den 15: 80 mg | 40 mg každý druhý týden |
Ulcerativní kolitida
Dospělí
Doporučená subkutánní dávka Humira u dospělých pacientů s ulcerózní kolitidou je 160 mg zpočátku v den 1 (poskytnuto za jeden den nebo rozděleno po dobu dvou po sobě jdoucích dnů), po které následuje 80 mg o dva týdny později (den 15). O dva týdny později (29. den) pokračují dávkováním 40 mg každý druhý týden.
Přerušte humiru u dospělých pacientů bez důkazu klinické remise o osm týdnů (57. den) terapie. Aminosalicyláty a/nebo kortikosteroidy mohou pokračovat během léčby Humirou. Azathioprin a 6-merkaptopurin (6-MP) [viz VAROVÁNÍS AND OPATŘENÍ ] může pokračovat během léčby Humirou v případě potřeby.
Pediatrie
Doporučená subkutánní dávka Humira u dětských pacientů ve věku 5 let a starší s ulcerózní kolitidou je založena na tělesné hmotnosti, jak je ukázáno níže:
| Pediatrická váha | Doporučené dávkování | |
| Dny až 15 | Počínaje 29. den* | |
| 20 kg (44 liber) na méně než 40 kg (88 liber) | Den 1: 80 mg den 8: 40 mg den 15: 40 mg | 40 mg každý druhý týden or 20 mg every week |
| 40 kg (88 liber) a větší | Den 1: 160 mg (jediná dávka nebo rozdělení po dobu dvou po sobě jdoucích dnů) 8: 80 mg den 15: 80 mg | 80 mg každý druhý týden nebo 40 mg každý týden |
| * Pokračujte v doporučené pediatrické dávce u pacientů, u kterých se stane 18 let a kteří jsou dobře kontrolovaní na jejich režimu Humira. |
Plakesová psoriáza Or Dospělý uveitida
Doporučená subkutánní dávka Humira u dospělých pacientů s plaktem psoriáza (PS) nebo Uveitis (UV) je počáteční dávka 80 mg, po které následuje 40 mg, který dává každý druhý týden od jednoho týdne po počáteční dávce. Použití humiry ve středním až závažném chronickém PS po jednom roce nebylo v kontrolovaných klinických studiích hodnoceno.
Hidradenitis Supsurative
Dospělí
Doporučená subkutánní dávka Humiry pro dospělé pacienty s hidradenitidou Supsurativa (HS) je počáteční dávka 160 mg (podávaná za jeden den nebo rozdělena po dobu dvou po sobě jdoucích dnů), po níž následuje 80 mg o dva týdny později (den 15). Začněte 40 mg týdně nebo 80 mg každý druhý týden dávkování o dva týdny později (29. den).
Adolescenti
Doporučená subkutánní dávka Humira u dospívajících pacientů ve věku 12 let a starší váží nejméně 30 kg s hidradenitidou Supsurativa (HS) je založena na tělesné hmotnosti, jak je uvedeno níže [viz viz [viz viz níže [Viz Použití v konkrétních populacích a Klinická farmakologie ]:
| Tělesná hmotnost dospívajících pacientů (12 let a starších) | Doporučené dávkování |
| 30 kg (66 liber) na méně než 60 kg (132 liber) |
|
| 60 kg (132 liber) a větší |
|
Monitorování pro posouzení bezpečnosti
Před zahájením humiry a pravidelného během terapie hodnotí pacienty na aktivní tuberkulózu a testu na latentní infekci [viz viz VAROVÁNÍS AND OPATŘENÍ ].
Obecné úvahy o správě
Humira je určena k použití pod vedením a dohledem lékaře. Pacient může vlastní injekci Humiru nebo pečovatel může vložit Humiru za použití buď humira pero nebo předplněné stříkačky, pokud lékař zjistí, že je vhodný a s lékařským sledováním podle potřeby po řádném tréninku v technice subkutánní injekce.
Humira může být vyřazena z ledničky po dobu 15 až 30 minut před vstřikováním, aby se kapalina dostala na teplotu místnosti. Neodstraňujte uzávěr ani kryt a zároveň jej nedovolte dosáhnout teploty místnosti. Pečlivě prohlédněte roztok v předplacené injekční injekční injekční lahvičce HUMIRA PREED PREED ANDEDUSE Institutional Use Vial pro subkulační hmotu a zbarvení. Pokud jsou zaznamenány částice a zbarvení, nepoužívejte produkt. Humira neobsahuje konzervační látky; proto zlikvidujte nevyužité části léčiva zbývajících z injekční stříkačky. Poznámka: Pokyn pacientům citlivým na latex, aby nezvládli kryt jehly Humira 40 mg/0,8 ml pera a 40 mg/0,8 ml 20 mg/0,4 ml a 10 mg/0,2 ml předplněné stříkačky, protože může obsahovat přírodní latex kaučuku [viz [viz [viz Jak dodáno / Skladování a manipulace ].
Poučte pacienty s používáním humira nebo předplněné stříkačky, aby vložili plné množství do stříkačky podle pokynů uvedených v pokynech pro použití [viz viz Pokyny pro použití ].
Injekce by měly nastat na samostatných místech ve stehně nebo břiše. Otočte místa vstřikování a nedávejte injekce do oblastí, kde je pokožka něžně pohmožděná červená nebo tvrdá.
Humira s jednodávkou institucionální používání je pro správu v institucionálním prostředí pouze jako je kancelář nebo klinika nemocnice. Získejte dávku pomocí sterilní jehly a injekční stříkačky a okamžitě se spravujte poskytovatelem zdravotní péče v institucionálním prostředí. Spravujte pouze jednu dávku na lahvičku. Lahvička neobsahuje konzervační látky; Proto zahoďte nepoužité porce.
Jak dodáno
Dávkování Forms And Strengths
Humira je jasné a bezbarvé řešení dostupné jako:
- Pen (HUMIRA PEN)
- Injekce: 80 mg/0,8 ml v pero s jednou dávkou.
- Injekce: 40 mg/0,8 ml v pero s jednou dávkou.
- Injekce: 40 mg/0,4 ml v pero s jednou dávkou.
- Předplněná stříkačka
- Injekce: 80 mg/0,8 ml v předplnivované skleněné stříkačce s jednou dávkou.
- Injekce: 40 mg/0,8 ml v jedné dávce předplněné skleněné stříkačce.
- Injekce: 40 mg/0,4 ml v předplnivované skleněné stříkačce s jednou dávkou.
- Injekce: 20 mg/0,4 ml v předplnivované skleněné stříkačce s jednou dávkou.
- Injekce: 20 mg/0,2 ml v jedné dávce předplněné skleněné stříkačky.
- Injekce: 10 mg/0,2 ml v jedné dávce předplněné skleněné stříkačky.
- Injekce: 10 mg/0,1 ml v předběžné skleněné stříkačce s jednou dávkou.
- Jednorázová institucionální použití lahvičky
- Injekce: 40 mg/0,8 ml v jednodávkové skleněné lahvičce pouze pro institucionální použití.
Skladování a manipulace
Humira® (Adalimumab) je dodáván jako sterilní a bezbarvý roztok bez konzervační látky pro subkutánní podání. K dispozici jsou následující konfigurace balení.
- Humira Pen Carton - 40 mg/0.8 mL
- Humira is supplied in a carton containing two alcohol preps a two dose trays. Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed ½ inch needle providing 40 mg/0.8 mL of Humira. The needle cover may contain natural třítber latex. The NDC Číslo je 0074-4339-02.
- Humira Pen Carton - 40 mg/0,4 ml
- Humira is supplied in a carton containing two alcohol preps a two dose trays. Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 40 mg/0,4 ml of Humira. The black needle cover is ne made with natural třítber latex. The NDC Číslo je 0074-0554-02.
- Humira Pen Carton †80 mg/0.8 mL
- Humira is supplied in a carton containing two alcohol preps a two dose trays. Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 80 mg/0.8 mL of Humira. The black needle cover is ne made with natural třítber latex. The NDC Číslo je 0074-0124-02.
- Humira Pen 40 mg/0.8 mL - Starter Package for Crohn's Disease Ulcerativní kolitida or Hidradenitis Supsurative
- Humira is supplied in a carton containing 6 alcohol preps a 6 dose trays (Starter Package for Ulcerativní kolitida or Hidradenitis Supsurative). Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed ½ inch needle providing 40 mg/0.8 mL of Humira. The needle cover may contain natural třítber latex. The NDC Číslo je 0074-4339-06.
- Humira Pen 40 mg/0,4 ml - Starter Package for Crohn's Disease Ulcerativní kolitida or Hidradenitis Supsurative
- Humira is supplied in a carton containing 6 alcohol preps a 6 dose trays (Starter Package for Ulcerativní kolitida or Hidradenitis Supsurative). Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 40 mg/0,4 ml of Humira. The black needle cover is ne made with natural třítber latex. The NDC Číslo je 0074-0554-06.
- Humira Pen 80 mg/0.8 mL - Starter Package for Crohn's Disease Ulcerativní kolitida or Hidradenitis Supsurative
- Humira is supplied in a carton containing 4 alcohol preps a 3 dose trays (Starter Package for Ulcerativní kolitida or Hidradenitis Supsurative). Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 80 mg/0.8 mL of Humira. The black needle cover is ne made with natural třítber latex. The NDC Číslo je 0074-0124-03.
- Humira Pen 40 mg/0.8 mL - Psoriasis Uveitida or Adolescent Hidradenitis Supsurative Starter Package
- Humira is supplied in a carton containing 4 alcohol preps a 4 dose trays (Psoriasis Uveitida or Adolescent Hidradenitis Supsurative Starter Package). Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed ½ inch needle providing 40 mg/0.8 mL of Humira. The needle cover may contain natural třítber latex. The NDC Číslo je 0074-4339-07.
- Humira Pen 40 mg/0,4 ml - Psoriasis Uveitida or Adolescent Hidradenitis Supsurative Starter Package
- Humira is supplied in a carton containing 4 alcohol preps a 4 dose trays (Psoriasis Uveitida or Adolescent Hidradenitis Supsurative Starter Package). Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 40 mg/0,4 ml of Humira. The black needle cover is ne made with natural třítber latex. The NDC Číslo je 0074-0554-04.
- Humira Pen 80 mg/0.8 mL a 40 mg/0,4 ml - Psoriasis Uveitida or Adolescent Hidradenitis Supsurative Starter Package
- Humira is supplied in a carton containing 4 alcohol preps a 3 dose trays (Psoriasis Uveitida or Adolescent Hidradenitis Supsurative Starter Package). One dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 80 mg/0.8 mL of Humira. The other two dose trays each consist of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 40 mg/0,4 ml of Humira. The black needle cover is ne made with natural třítber latex. The NDC Číslo je 0074-1539-03.
- Humira Pen 80 mg/0.8 mL †Starter Package for Pediatrická ulcerózní kolitida (4 count)
- Humira is supplied in a carton containing 4 alcohol preps a 4 dose trays (Starter Package for Pediatrická ulcerózní kolitida). Each dose tray consists of a single-dose pen containing a 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 80 mg/0.8 mL of Humira. The black needle cover is ne made with natural třítber latex. The NDC Číslo je 0074-0124-04.
- Předplněná stříkačka Carton - 40 mg/0.8 mL
- Humira is supplied in a carton containing two alcohol preps a two dose trays. Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed ½ inch needle providing 40 mg/0.8 mL of Humira. The needle cover may contain natural třítber latex. The NDC Číslo je 0074-3799-02.
- Předplněná stříkačka Carton - 40 mg/0,4 ml
- Humira is supplied in a carton containing two alcohol preps a two dose trays. Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 40 mg/0,4 ml of Humira. The black needle cover is ne made with natural třítber latex. The NDC Číslo je 0074-0243-02.
- Předplněná stříkačka Carton - 20 mg/0.4 mL
- Humira is supplied in a carton containing two alcohol preps a two dose trays. Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed ½ inch needle providing 20 mg/0.4 mL of Humira. The needle cover may contain natural třítber latex. The NDC Číslo je 0074-9374-02.
- Předplněná stříkačka Carton - 20 mg/0.2 mL
- Humira is supplied in a carton containing two alcohol preps a two dose trays. Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 20 mg/0.2 mL of Humira. The black needle cover is ne made with natural třítber latex. The NDC Číslo je 0074-0616-02.
- Předplněná stříkačka Carton - 10 mg/0.2 mL
- Humira is supplied in a carton containing two alcohol preps a two dose trays. Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed ½ inch needle providing 10 mg/0.2 mL of Humira. The needle cover may contain natural třítber latex. The NDC Číslo je 0074-6347-02.
- Předplněná stříkačka Carton - 10 mg/0.1 mL
- Humira is supplied in a carton containing two alcohol preps a two dose trays. Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 10 mg/0.1 mL of Humira. The black needle cover is ne made with natural třítber latex. The NDC Číslo je 0074-0817-02.
- Humira Předplněná stříkačka 40 mg/0.8 mL - Pediatrická Crohnova nemoc Starter Package (6 count)
- Humira is supplied in a carton containing 6 alcohol preps a 6 dose trays (Pediatric Starter Package). Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed ½ inch needle providing 40 mg/0.8 mL of Humira. The needle cover may contain natural třítber latex. The NDC Číslo je 0074-3799-06.
- Humira Předplněná stříkačka 80 mg/0.8 mL - Pediatrická Crohnova nemoc Starter Package (3 count)
- Humira is supplied in a carton containing 4 alcohol preps a 3 dose trays (Pediatric Starter Package). Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 80 mg/0.8 mL of Humira. The black needle cover is ne made with natural třítber latex. The NDC Číslo je 0074-2540-03.
- Humira Předplněná stříkačka 40 mg/0.8 mL - Pediatrická Crohnova nemoc Starter Package (3 count)
- Humira is supplied in a carton containing 4 alcohol preps a 3 dose trays (Pediatric Starter Package). Each dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed ½ inch needle providing 40 mg/0.8 mL of Humira. The needle cover may contain natural třítber latex. The NDC Číslo je 0074-3799-03.
- Humira Předplněná stříkačka 80 mg/0.8 mL a 40 mg/0,4 ml - Pediatrická Crohnova nemoc Starter Package (2 count)
- Humira is supplied in a carton containing 2 alcohol preps a 2 dose trays (Pediatric Starter Package). One dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 80 mg/0.8 mL of Humira. The other dose tray consists of a single-dose 1 mL prefilled glass syringe with a fixed thin wall ½ inch needle providing 40 mg/0,4 ml of Humira. The black needle cover is ne made with natural třítber latex. The NDC Číslo je 0074-0067-02.
- Jednorázová institucionální použití lahvičky Carton - 40 mg/0.8 mL
- Humira is supplied for institutional use only in a carton containing a single-dose glass vial providing 40 mg/0.8 mL of Humira. The vial stopper is ne made with natural třítber latex. The NDC
Skladování a stabilita
Nepoužívejte po datu vypršení platnosti na kontejneru. Humira musí být chlazena při 36 ° F až 46 ° F (2 ° C až 8 ° C). Ne zmrazení. Nepoužívejte, pokud je zmrazen, i když byl rozmrazen.
Uložte v původním kartonu až do doby správy, abyste chránili před světlem.
V případě potřeby může být například při cestování Humira uložena při teplotě místnosti až do maximálně 77 ° F (25 ° C) po dobu až 14 dnů s ochranou před světlem. Humira by měla být vyřazena, pokud se nepoužívá během 14denního období. Zaznamenejte datum, kdy je Humira poprvé odstraněna z chladničky v prostorech uvedených na kartonu a dávce.
Neukládejte humiru v extrémním teplu nebo chladu.
ABBVIE Inc. North Chicago IL 60064 USA. Revidováno: únor 2021
Vedlejší účinky for Humira
Následující klinicky významné nežádoucí účinky jsou popsány jinde při označování:
- Vážné infekce [see VAROVÁNÍS AND OPATŘENÍ ]
- Malignity [viz VAROVÁNÍS AND OPATŘENÍ ]
- Reakce přecitlivělosti [viz VAROVÁNÍS AND OPATŘENÍ ]
- Reaktivace viru hepatitidy B [viz VAROVÁNÍS AND OPATŘENÍ ]
- Neurologické reakce [viz VAROVÁNÍS AND OPATŘENÍ ]
- Hematologické reakce [viz VAROVÁNÍS AND OPATŘENÍ ]
- Srdeční selhání [viz VAROVÁNÍS AND OPATŘENÍ ]
- Autoimunity [viz VAROVÁNÍS AND OPATŘENÍ ]
Zkušenosti z klinických studií
Protože klinické studie se provádějí za široce proměnlivých podmínek, nežádoucí rychlosti nežádoucí reakce pozorované v klinických studiích léčiva nelze přímo porovnat s mírami v klinických studiích s jiným lékem a nemusí odrážet míru pozorované v praxi.
Nejběžnější nežádoucí reakcí s Humirou byly reakce injekce. V průběhu průchodů se v průběhu placebokontrolovaných 20% pacientů léčilo reakce v injekčním místě (erytém a/nebo svědění bolesti krvácení nebo otoky) ve srovnání se 14% pacientů, kteří dostávají placebo. Většina reakcí místa injekce byla popsána jako mírná a obecně nevyžadovala přerušení léčiva.
Podíl pacientů, kteří ukončili léčbu v důsledku nežádoucích účinků během dvojitě slepé placebem kontrolované části studií u pacientů s RA (tj. Studie Ra-I Raii RA-III a RA-IV), byl 7% u pacientů užívajících HUMIRA a 4% u pacientů s placebem. Nejčastějšími nežádoucími účinky vedoucími k přerušení Humira v těchto studiích RA byly vyrážky z klinické erupce (NULL,7%) (NULL,3%) a pneumonie (NULL,3%).
Infekce
V kontrolovaných částech 39 globálních klinických studií Humira u dospělých pacientů s RA PSA jako CD UC PS HS a UV byla míra závažných infekcí 4,3 na 100 pacientů u 7973 pacientů ošetřených humirou oproti míru 2,9 na 100 pacientů u 4848 pacientů s kontrolou. Pozorované vážné infekce zahrnovaly pneumonii septická artritida protetická a posturgická infekce erysipelas celulitida divertikulitida a pyelonefritida [viz viz VAROVÁNÍS AND OPATŘENÍ ].
Tuberkulóza a oportunní infekce
U 52 globálních kontrolovaných a nekontrolovaných klinických studií v RA PSA jako CD UC PS HS a UV, které zahrnovaly 24605 pacientů ošetřených humirou, byla míra hlášené aktivní tuberkulózy 0,20 na 100 pacientů a byl pozitivní přeměnu PPD 0,09 na 100 pacientových let. V podskupině 10113 pacientů ošetřených americkými a kanadskými humirami byla míra hlášených aktivních TB 0,05 na 100 pacientů a míra pozitivní konverze PPD byla 0,07 na 100 pacientů. Tyto studie zahrnovaly zprávy o miliarně lymfatické peritoneální a plicní TB. VAROVÁNÍS AND OPATŘENÍ ].
Autoprotilátky
V revmatoidní artritidě kontrolovaly studie 12% pacientů léčených Humirou a 7% pacientů ošetřených placebem, kteří měli negativní základní titry ANA, vyvinuly pozitivní titry ve 24. týdnu. Dva pacienti z 3046 léčených humirou vyvinuli klinické příznaky syndromu podobného Newonset Lupus -like. Po ukončení léčby se pacienti zlepšili. Žádní pacienti se nevyvinuli lupusovou nefritidu nebo symptomy centrálního nervového systému. Dopad dlouhodobé léčby Humirou na vývoj autoimunitních onemocnění není znám.
Zvýšení jaterního enzymu
Zprávy o závažných jaterních reakcích, včetně akutního selhání jater u pacientů, kteří dostávali blokátory TNF. V kontrolované fázi 3 studií HUMIRA (40 mg SC každý druhý týden) u pacientů s RA PSA a stejně jako u doby kontrolního období v rozmezí od 4 do 104 týdnů ALT zvýšení ≥ 3 x Uln se vyskytlo u 3,5% pacientů ošetřených HUMIRA a 1,5% pacientů s kontrolou. Protože mnoho z těchto pacientů v těchto studiích užívalo také léky, které způsobují zvýšení enzymu jater (např. NSAIDS MTX), vztah mezi humirou a zvýšením enzymu jaterní není jasný. V kontrolované fázi 3 studie HUMIRA u pacientů s polyartikulární JIA, kteří byli 4 až 17 let ALT zvýšení ≥ 3 x ULN se vyskytovaly u 4,4% pacientů ošetřených humirou a 1,5% pacientů s kontrolou (alt běžnější než AST); Zvýšení testu jaterního enzymu bylo častější mezi těmi, které byly ošetřeny kombinací Humiry a MTX, než je ošetřené samotnou humirou. Obecně tato výška nevedla k přerušení humirské léčby. Žádné výšky ALT ≥ 3 x Uln se nevyskytovalo v otevřené studii HUMIRA u pacientů s polyartikulární jia, kteří byli 2 až <4 years.
V kontrolované fázi 3 studií Humira (počáteční dávky 160 mg a 80 mg nebo 80 mg a 40 mg ve dnech 1 a 15, po kterém následuje 40 mg každý druhý týden) u dospělých pacientů s Crohnovou chorobou s kontrolním obdobími v rozmezí od 4 do 52 týdnů ALT výšky, se vyskytovaly u pacientů s 0,9%, které se chovaly hladiny pacientů. Ve fázi 3 studie HUMIRA u pediatrických pacientů s Crohnovou chorobou, která hodnotila účinnost a bezpečnost dvou režimů založené na tělesné hmotnosti po údržbě po indukční terapii založené na tělesné hmotnosti až do 52 týdnů léčby ≥ 3 x ULN se vyskytly u 2,6% (5/192), z nichž 4 se dostávali souběžné imunosupresivy; Žádný z těchto pacientů neukončil kvůli abnormalitám v ALT testech. V kontrolované fázi 3 studií HUMIRA (počáteční dávky 160 mg a 80 mg ve dnech 1 a 15, po kterém následuje 40 mg každý druhý týden) u dospělých pacientů s UC s dobou kontroly v průběhu 1 do 52 týdnů alt výšky ≥ 3 x ULN se vyskytovaly u 1,5% pacientů ošetřených HUMIRA. V kontrolované fázi 3 studie HUMIRA u pacientů s dětskou ulcerózní kolitidou (n = 93), která hodnotila účinnost a bezpečnost udržovací dávky 0,6 mg/kg (maximálně 40 mg) každý druhý týden (n = 31) a údržbářská dávka (maximálně 40 mg) každý týden každý týden (n = 32) po tělesnou hmotnost mg) v týdnu 0 a týden 1 a 1,2 mg/kg (maximálně 80 mg) v týdnu 2 (n = 63) nebo indukční dávce 2,4 mg/kg (maximálně 160 mg) v týdnu 0 placeba v 1 a 1,2 mg/kg (maximum 80 mg) ve 2. týdnu (n = 30) ALT se vyskytovala u 1,1% (1 1/93) (1 1/93) (1/93) (1/93). V kontrolované fázi 3 studií HUMIRA (počáteční dávka 80 mg, pak 40 mg každý druhý týden) u pacientů s PS s dobou kontroly v rozmezí od 12 do 24 týdnů ALT zvýšení ≥ 3 x Uln se vyskytlo u 1,8% pacientů ošetřených humirou a 1,8% pacientů s kontrolou. V kontrolovaných pokusech s humirou (počáteční dávky 160 mg v týdnu 0 a 80 mg ve 2. týdnu následované 40 mg každý týden počínaje 4. týdnem) u subjektů s HS s dobou kontroly v rozmezí od 12 do 16 týdnů alt výšky ≥ 3 x ULN se vyskytovalo u 0,3% humora ošetřených subjektů a 0,6% subjektů s kontrolou. V kontrolovaných studiích s humirou (počáteční dávky 80 mg v 0. týdnu následované 40 mg každý druhý týden počínaje 1. týdnem) u dospělých pacientů s uveitidou s expozicí 165,4 Pys a 119,8 Pys u humira ošetřených a kontrolních pacientů se vyskytovalo u pacientů s hladinou a u pacientů s hladinou.
Jiné nežádoucí účinky
Revmatoidní artritida Klinické studie
Níže popsané údaje odrážejí expozici Humiru u 2468 pacientů včetně 2073 exponovaných po dobu 6 měsíců 1497 vystavených po dobu delší než jeden rok a 1380 v přiměřených a dobře kontrolovaných studiích (studie Ra-I Ra-II RA-III a RA-IV). Humira byla studována primárně v placebokontrolních studiích a v dlouhodobých sledovacích studiích po dobu až 36 měsíců. Populace měla průměrný věk 54 let 77% žen 91% byly Kavkazské a mírně měly vážně aktivní revmatoidní artritidu. Většina pacientů dostávala 40 mg humira každý druhý týden [viz Klinické studie ].
Tabulka 1 shrnuje reakce uváděné rychlostí nejméně 5% u pacientů léčených Humirou 40 mg každý druhý týden ve srovnání s placebem as výskytem vyšší než placebo. Ve studii RA-III byly typy a frekvence nežádoucích účinků ve druhém ročníku prodloužení s otevřeným bodem podobné frekvenci pozorovaným v jednoleté dvojitě slepé části.
Tabulka 1: Nežádoucí účinky hlášené ≥ 5% pacientů léčených Humirou během placebem kontrolovaného období sdružených RA studií (studie RA-I Ra-II RA-III a RA-IV)
| Humira 40 mg subcutaneous Every Ostatní Week (N = 705) | Placebo (N = 690) | |
| Nežádoucí reakce (preferovaný termín) | ||
| Respirační | ||
| Infekce horních cest dýchacích | 17% | 13% |
| Sinusitida | 11% | 9% |
| Chřipkový syndrom | 7% | 6% |
| Gastrointestinal | ||
| Nevolnost | 9% | 8% |
| Bolest břicha | 7% | 4% |
| Laboratorní testy* | ||
| Laboratorní test abnormální | 8% | 7% |
| Hypercholesterolemie | 6% | 4% |
| Hyperlipidemie | 7% | 5% |
| Hematurie | 5% | 4% |
| Alkalická fosfatáza se zvýšila | 5% | 3% |
| Ostatní | ||
| Bolest hlavy | 12% | 8% |
| Vyrážka | 12% | 6% |
| Náhodné zranění | 10% | 8% |
| Reakce místa injekce ** | 8% | 1% |
| Bolest zad | 6% | 4% |
| Infekce močových cest | 8% | 5% |
| Hypertenze | 5% | 3% |
| * Abnormality laboratorních testů byly hlášeny jako nežádoucí účinky v evropských studiích ** nezahrnuje injekční místo Erythema svědění bolesti krvácení nebo otoky |
Méně běžné nežádoucí účinky v klinických studiích revmatoidní artritidy
Ostatní infrequent serious adverse reactions that ne appear in the Warnings a Precautions or Adverse Reaction sections that occurčervený at an incidence of less than 5% in Humira-treated patients in RA studies were:
Tělo jako celek: Bolest v končetiny operace pánevní bolesti
Kardiovaskulární systém: Arytmie Fibrilation Bolest na hrudi Koronární tepna Porucha srdeční zástava Hypertenzní encefalopatie myokardiální infarkt Palpitace Perikardiální výtok Perikarditida synkop tachykardie tachykardie
Trávicí systém: Cholecystitida cholelitiáza ezofagitida Gastrointeritida Gastrointestinální krvácení Jaterní nekróza zvracení zvracení
Endokrinní systém: Porucha parathyroidy
Hemický a lymfatický systém: Polycytémie agranulocytózy
Metabolické a nutriční poruchy: Dehydratační hojení abnormální ketózy Paraproteinemie Periferní edém
Musculo -skeletal System: Artritida kostní porucha zlomeniny kosti (ne spontánní) kostní nekróza kloubní porucha svalové křeče myasthenia pyogenní artritida synovitida porucha šlachy šlachy
Neoplasie: Adenom
Nervový systém: Zmatek Parestezie subdurální hematomový tremor
Respirační System: Astma Bronchospasm Dyspnea plicní funkce snížila pleurální výpotek
Speciální smysly: Šedý zákal
Trombóza: Noha trombózy
Urogenitální systém: Cystitida ledviny počítá menstruační porucha
Juvenilní idiopatická artritida Klinické studie
Obecně nežádoucí účinky u pacientů ošetřených humirou v pokusech o polyartikulární juvenilní idopathis artritidu (JIA) (studie JIA-I a JIA-II) [viz viz Klinické studie ] byly podobné frekvenci a typu jako u dospělých pacientů [viz VAROVÁNÍS AND OPATŘENÍ Nežádoucí účinky ]. Important findings a differences from adults are discussed in the following paragraphs.
Ve studii byl JIA-I HUMIRA studován u 171 pacientů, u kterých bylo 4 až 17 let s polyartikulární JIA. Závažné nežádoucí účinky hlášené ve studii zahrnovaly streptokokovou faryngitidu neutropenie zvýšené aminotransferázy herpes zoster myositis metrorrhagia a apendicitidu. Vážné infekce byly pozorovány u 4% pacientů do přibližně 2 let od zahájení léčby Humirou a zahrnovaly případy herpes simplex pneumonia močového traktu infekce faryngitidy a herpes zoster.
Ve studii se Jia-I 45% pacientů zažilo infekci při dostávání Humira s nebo bez doprovodných MTX v prvních 16 týdnech léčby. Typy infekcí hlášených u pacientů ošetřených humirou byly obecně podobné těm, které jsou běžně pozorovány u pacientů s polyartikulárními jia, kteří nejsou léčeni blokátory TNF. Po zahájení léčby byly nejčastějšími nežádoucími účinky, ke kterým došlo v této populaci pacientů léčené humirou, bolest v místě injekce a reakce místa injekce (19% a 16%). Méně běžně hlášenou nežádoucí příhodou u pacientů, kteří dostávali humiru, byl granulom annulare, který nevedl k přerušení humirské léčby.
V prvních 48 týdnech léčby ve studii byly u u 6% pacientů pozorovány reakce na nesmyslivosti JIA-I, které nejsou vážné přecitlivělosti a zahrnovaly primárně lokalizované alergické hypersenzitivní reakce a alergické vyrážky.
Ve studii jia-I 10% pacientů léčených Humirou, kteří měli negativní základní anti-dsDNA protilátky po 48 týdnech léčby. Během klinického hodnocení žádný pacient nevyvinul klinické příznaky autoimunity.
Přibližně 15% pacientů léčených Humirou se vyvinulo mírnou až střední zvýšení kreatinu fosfokinázy (CPK) ve studii Jia-I. U několika pacientů byla pozorována zvýšení přesahující pětinásobek horní hranice normálu. Koncentrace CPK se u všech pacientů snížily nebo se vrátily k normálu. Většina pacientů byla schopna pokračovat v Humiru bez přerušení.
Ve studii byl JIA-II HUMIRA studován u 32 pacientů, kteří byli 2 až <4 years of age or 4 years of age a older weighing <15 kg with polyarticular JIA. The safety profile for this patient population was similar to the safety profile seen in patients 4 až 17 let with polyarticular JIA.
Ve studii jia-II 78% pacientů zažilo infekci při přijímání Humiry. Mezi ně patřili infekce horní cest dýchacích cest nasopharyngitis bronchitida a byly většinou mírné až střední závažnosti. Vážné infekce byly pozorovány u 9% pacientů, kteří ve studii dostávali Humiru, a zahrnovali rotavirusový gastroenteritidu a varicelu zubního kazu.
Ve studii byly u 6% pacientů pozorovány nesmírné alergické reakce JIA-II a zahrnovaly přerušovanou urticizaci a vyrážku, které byly v závažnosti mírné.
Psoriatická artritida And Ankylozující spondylitida Klinické studie
Humira has been studied in 395 patients with psoriatic artritida (PsA) in two placebocontrolled trials a in an open label study a in 393 patients with ankylosing spondylitis (AS) in two placebo-controlled studies [see Klinické studie ]. The safety profile for patients with PsA a AS treated with Humira 40 mg každý druhý týden was similar to the safety profile seen in patients with RA Humira Studies RA-I through IV.
Klinické studie Crohnovy choroby
Dospělí
Bezpečnostní profil Humira u 1478 dospělých pacientů s Crohnovou chorobou ze čtyř placebem kontrolovaných a dvou studií prodloužení s otevřenou značkou [viz viz Klinické studie ] byl podobný bezpečnostnímu profilu pozorovanému u pacientů s RA.
Pediatričtí pacienti 6 až 17 let
Bezpečnostní profil HUMIRA v roce 192 Pediatrických pacientů z jedné dvojitě zaslepené studie (studie PCD-I) a jedné studie s otevřeným znakem [viz viz Klinické studie ] byl podobný bezpečnostnímu profilu pozorovanému u dospělých pacientů s Crohnovou chorobou. Během 4týdenní fáze indukce otevřené značky studie PCD-I Nejběžnější nežádoucí účinky vyskytující se v dětské populaci léčené humirou byly bolesti v místě injekce a reakce místa injekce (6% a 5%).
Celkem 67% dětí zažilo infekci při přijímání Humiry ve studii PCD-I. Jednalo se o infekci horních cest dýchacích a nasofaryngitidy.
Celkem 5% dětí zažilo vážnou infekci při přijímání Humiry ve studii PCD-I. Jednalo se o sepsis sepse virového infekce (katétru) gastroenteritida H1N1 chřipku a diseminovaná histoplazmóza.
Ve studii byly však alergické reakce PCD-I pozorovány u 5% dětí, které byly všechny nesmírné a byly primárně lokalizované reakce.
Ulcerativní kolitida Klinické studie
Dospělí
Bezpečnostní profil HUMIRA u 1010 dospělých pacientů s ulcerózní kolitidou (UC) ze dvou placebem kontrolovaných studií a jednou studií prodloužení s otevřenou značkou [viz viz Klinické studie ] byl podobný bezpečnostnímu profilu pozorovanému u pacientů s RA.
Pediatričtí pacienti 5 až 17 let
Bezpečnostní profil Humira u 93 pediatrických pacientů s ulcerativní kolitidou z jedné dvojitě zaslepené studie a jedné studii prodloužení s otevřenou značkou [viz viz Klinické studie ] byl podobný bezpečnostnímu profilu pozorovanému u dospělých pacientů s ulcerativní kolitidou.
Plakesová psoriáza Klinické studie
Humira has been studied in 1696 subjects with plaque psoriasis (Ps) in placebo-controlled a open-label extension studies [see Klinické studie ]. The safety profile for subjects with Ps treated with Humira was similar to the safety profile seen in subjects with RA with the following exceptions. In the placebo-controlled portions of the clinical trials in Ps subjects Humira-treated subjects had a higher incidence of arthralgia when compačervený to controls (3% vs. 1%).
Hidradenitis Supsurative Klinické studie
Humira has been studied in 727 subjects with hidradenitis suppurativa (HS) in three placebocontrolled studies a one open-label extension study [see Klinické studie ]. The safety profile for subjects with HS treated with Humira weekly was consistent with the known safety profile of Humira.
Vzplanutí HS definované jako ≥ 25% se zvýšilo z výchozí hodnoty u abscesů a počtu zánětlivých uzlů a s minimálně 2 dalšími lézemi byl zdokumentován u 22 (22%) ze 100 subjektů, kteří byli staženi z léčby HUMIRA po primárním časové účinku ve dvou studiích.
Uveitida Klinické studie
Humira has been studied in 464 adult patients with uveitis (UV) in placebo-controlled a open-label extension studies a in 90 pediatric patients with uveitis (Studujte puv-i) [see Klinické studie ]. Bezpečnostní profil u pacientů s UV léčeným humirou byl podobný bezpečnostnímu profilu pozorovanému u pacientů s RA.
Imunogenita
Stejně jako u všech terapeutických proteinů existuje potenciál pro imunogenitu. Detekce tvorby protilátek je vysoce závislá na citlivosti a specificitě testu. Pozorovaný výskyt protilátky (včetně neutralizační protilátky) pozitivity v testu může být navíc ovlivněn několika faktory, včetně metodiky pro testovací metodiku, která manipuluje načasování vzorku doprovodných léků a základní onemocnění. Z těchto důvodů může být porovnání výskytu protilátek ve studiích popsaných s výskytem protilátek v jiných studiích nebo na jiných produktech adalimumab zavádějící.
Existují dva testy, které byly použity k měření anti-adalimumab protilátek. S protilátkami ELISA k Adalimumabu mohly být detekovány pouze tehdy, když byly koncentrace séra adalimumab <2 mcg/mL. The ECL assay can detect anti-Adalimumab antibody titers independent of Adalimumab concentrations in the serum samples. The incidence of antiAdalimumab antibody (AAA) development in patients treated with Humira are presented in Table 2.
Tabulka 2: Vývoj protilátky proti adalimumabu určený ELISA a testem ECL u pacientů léčených Humirou
| Indikace | Doba trvání studie | Výskyt protilátky proti adalimumabu pomocí ELISA (N/N) | Výskyt protilátky proti adalimumabu pomocí testu ECL (N/N) | ||
| U všech pacientů, kteří dostávali adalimumab | U pacientů s koncentracemi séra adalimumab <2 mcg/mL | ||||
| Revmatoidní artritida a | 6 až 12 měsíců | 5% (58/1062) | Žádný. | Na | |
| Juvenilní idiopatická artritida (JIA) | 4 až 17 let b | 48 týdnů | 16% (27/171) | Žádný. | Na |
| 2 až 4 roky nebo ≥ 4 roky věku a vážení <15 kg | 24 týdnů | 7% (1/15) c | Žádný. | Na | |
| Psoriatická artritida d | 48 týdnů e | 13% (24/178) | Žádný. | Na | |
| Ankylozující spondylitida | 24 týdnů | 9% (16/185) | Žádný. | Na | |
| Crohnova nemoc dospělého | 56 týdnů | 3% (7/269) | 8% (7/86) | Na | |
| Pediatrická Crohnova choroba | 52 týdnů | 3% (6/182) | 10% (6/58) | Na | |
| Ulcerativní kolitida dospělých | 52 týdnů | 5% (19/360) | 21% (19/92) | Na | |
| Pediatrická ulcerózní kolitida | 52 týdnů | 3% (3/100) | 13% (3/23) | 33% (33/100) i | |
| Plakesová psoriáza f | Až 52 týdnů g | 8% (77/920) | 21% (77/372) | Na | |
| Hidradenitis Supsurative | 36 týdnů | 7% (30/461) | 28% (58/207) h | 61% (272/445) j | |
| Neinfekční uveitida | 52 týdnů | 5% (12/249) | 21% (12/57) | 40% (99/249) k | |
| N: Počet pacientů s anti-adalimumab protilátkou; NR: Není hlášeno; NA: Nelze použít (neprováděno) a U pacientů, kteří dostávali souběžný methotrexát (MTX) b U pacientů, kteří dostávali souběžnou MTX, byl výskyt protilátky proti adalimumabu 6% ve srovnání s 26% s monoterapií Humira c Tento pacient obdržel souběžný MTX d U pacientů, kteří dostávali souběžný MTX, byl výskyt vývoje protilátek 7% ve srovnání s 1% v RA e Subjekty se zapsaly po dokončení 2 předchozích studií 24 týdnů nebo 12 týdnů léčby. f V plaku pacienti s psoriázou, kteří byli na monoterapii HUMIRA a následně staženi z léčby, byla míra protilátek na adalimumab po opakování podobná míře pozorované před stažením g Jedna 12týdenní studie fáze 2 a jedna 52týdenní studie fáze 3 h Mezi subjekty ve dvou studiích fáze 3, které zastavily léčbu Humirou po dobu až 24 týdnů a u nichž hladiny adalimumab séra následně poklesly <2 mcg/mL (approximately 22% of total subjects studied) i Nebyla pozorována žádná zjevná souvislost mezi vývojem protilátky a bezpečností. Asociace vývoje protilátek a výsledku účinnosti nebylo hodnoceno kvůli omezenému počtu subjektů v každé skupině léčené stratifikované anti-adalimumab protilátkovým titrem. j Nebyla pozorována žádná zjevná souvislost mezi vývojem protilátky a bezpečností k Nebyla pozorována žádná korelace vývoje protilátek na bezpečnost nebo výsledky účinnosti |
Revmatoidní artritida And Psoriatická artritida
Pacienti ve studiích RA-I RA-II a RA-III byli testováni ve více časových bodech pro protilátky vůči Adalimumabu pomocí ELISA během 6 až 12 měsíců. Nebyla pozorována žádná zjevná korelace vývoje protilátky na nežádoucí účinky. U pacientů s monoterapií, kteří dostávají každý druhý týden, se dávkování může vyvíjet protilátky častěji než u pacientů, kteří dostávají týdenní dávkování. U pacientů, kteří dostávali doporučenou dávkování 40 mg každý druhý týden, protože monoterapie byla reakce ACR 20 u pacientů pozitivních na protilátku nižší než u pacientů s protilátkou. Dlouhodobá imunogenita humiry není známa.
Zážitek z postmarketingu
Během použití Humira byly identifikovány následující nežádoucí účinky. Protože tyto reakce jsou hlášeny dobrovolně z populace nejisté velikosti, není vždy možné spolehlivě odhadnout jejich frekvenci nebo vytvořit kauzální vztah k expozici Humiru.
Gastrointestinal disorders: Divertikulitida Velká střevní perforace včetně perforací spojených s divertikulitidou a apendiceálními perforacemi spojenými s apendicitis pankreatitida
Obecné poruchy a podmínky správy na místě: Pyrexia
HEPATO-BILIÁLNÍ PORUŠENÍ: Selhání jater hepatitidy
Poruchy imunitního systému: Sarkoidóza
Neoplazmy benigní maligní a nespecifikované (včetně cyst a polypů): Karcinom Merkelových buněk (neuroendokrinní karcinom kůže)
Poruchy nervového systému: Demyelinizační poruchy (např. Syndrom optické neuritidy Guillain-Barré) cerebrovaskulární nehoda
Respirační disorders: Intersticiální onemocnění plic včetně plicní fibrózy plicní embolie
Kožní reakce: Stevens Johnsonův syndrom kožní vaskulitida Erythema Multiforme Nová nebo zhoršující se psoriáza (všechny podtypy včetně pustulárního a palmoplantaru) alopecie lichenoidní kožní reakce
Cévní poruchy: Systémová vaskulitida Trombóza hluboká žilní
Lékové interakce for Humira
Methotrexát
Humira has been studied in rheumatoid artritida (RA) patients taking concomitant methotrexate (MTX). Although MTX červenýuced the apparent Adalimumab clearance the data ne suggest the need for dose adjustment of either Humira or MTX [see Klinická farmakologie ].
Biologické produkty
V klinických studiích u pacientů s RA bylo pozorováno zvýšené riziko závažných infekcí s kombinací blokátorů TNF s Anakinrou nebo abataceptem bez přidané přínosy; U pacientů s RA se proto nedoporučuje používání Humira s abataceptem nebo Anakinrou [viz VAROVÁNÍS AND OPATŘENÍ ]. A higher rate of serious infections has also been observed in patients with RA treated with rituximab who received subsequent treatment with a TNF blocker. There is insufficient information regarding the concomitant use of Humira a other biologic products for the treatment of RA PsA AS CD UC Ps HS a UV. Concomitant administration of Humira with other biologic DMARDS (e.g. anakinra a abatacept) or other TNF blockers is ne recommended based upon the possible increased risk for infections a other potential pharmacological interactions.
Živé vakcíny
Vyhněte se použití živých vakcín s humirou [viz VAROVÁNÍS AND OPATŘENÍ ].
Substráty cytochromu P450
Tvorba enzymů CYP450 může být potlačena zvýšenými koncentracemi cytokinů (např. TNFa IL-6) během chronického zánětu. Je možné, že molekula antagonizuje aktivitu cytokinu, jako je adalimumab, ovlivňují tvorbu enzymů CYP450. Po zahájení nebo přerušení humir u pacientů léčených substráty CYP450 s úzkým monitorováním terapeutického indexu účinku (např. Warfarin) nebo koncentraci léčiva (např. Cyklosporin nebo Theofylin) a podle potřeby se doporučuje jednotlivá dávka léčivého produktu.
Varování pro Humiru
Zahrnuto jako součást OPATŘENÍ sekce.
Opatření pro Humiru
Vážné infekce
Pacienti léčeni Humirou jsou vystaveni zvýšenému riziku rozvoje závažných infekcí zahrnujících různé orgánové systémy a místa, které mohou vést k hospitalizaci nebo smrti. Oportunistické infekce způsobené bakteriální mykobakteriální invazivní plísňovou virovou parazitární nebo jinými oportunistickými patogeny včetně aspergilózy blastomykózy kandidóza kokcidioidomykózy histoplazmóza legionelózy listeriózy pneumocystosis pneumocystosis pneumocystosis. Pacienti často prezentovali diseminované spíše než lokalizované onemocnění.
Současné použití blokátoru TNF a abataceptu nebo anakinry bylo spojeno s vyšším rizikem vážných infekcí u pacientů s revmatoidní artritidou (RA); Současné použití Humiry a těchto biologických produktů se proto nedoporučuje při léčbě pacientů s RA [viz viz VAROVÁNÍS AND OPATŘENÍ a Lékové interakce ].
Léčba Humirou by neměla být zahájena u pacientů s aktivní infekcí včetně lokalizovaných infekcí. Pacienti ve věku 65 let a starší pacienti s komorbidními stavy a/nebo pacienti, kteří užívají doprovodné imunosupresivy (jako jsou kortikosteroidy nebo methotrexát), mohou být vystaveny většímu riziku infekce. Zvažte rizika a přínosy léčby před zahájením terapie u pacientů:
- s chronickou nebo opakující se infekcí;
- kteří byli vystaveni tuberkulóze;
- s historií oportunistické infekce;
- kteří pobývali nebo cestovali v oblastech endemické tuberkulózy nebo endemických mykosů, jako je histoplazmóza kokcidioidomykóza nebo blastomykóza; nebo
- za základních podmínek, které je mohou předisponovat k infekci.
Tuberkulóza
U pacientů, kteří dostávali Humiru, byly hlášeny případy reaktivace tuberkulózy a nových infekcí tuberkulózy tuberkulózy, včetně pacientů, kteří dříve dostávali léčbu latentní nebo aktivní tuberkulózy. Zprávy zahrnovaly případy plicní a extrapulmonální (tj. Diseminované) tuberkulózy. Vyhodnoťte pacienty na rizikové faktory tuberkulózy a testujte latentní infekci před zahájením humiry a pravidelně během terapie.
Bylo prokázáno, že léčba infekce latentní tuberkulózy před terapií blokovacími látkami TNF snižuje riziko reaktivace tuberkulózy během terapie. Před zahájením HUMIRA posoudí, zda je nutná léčba latentní tuberkulózy; a považujte induraci ≥ 5 mm za pozitivní výsledek kožního testu tuberkulinu i u pacientů dříve očkovaných Bacille Calmethe-Guerinem (BCG).
Zvažte terapii anti-tuberkulózy před zahájením humiry u pacientů s minulou anamnézou latentní nebo aktivní tuberkulózy, u níž nelze potvrdit přiměřený průběh léčby, a u pacientů s negativním testem na latentní tuberkulózu, ale s rizikovými faktory pro infekci tuberkulózy. Navzdory profylaktické léčbě případů tuberkulózy došlo u pacientů léčených Humirou. Konzultace s lékařem s odbornými znalostmi v oblasti léčby tuberkulózy se doporučuje pomoci při rozhodování, zda je pro individuálního pacienta vhodná zahájení terapie antituberkulózy.
Silně zvažte tuberkulózu u diferenciální diagnózy u pacientů, u nichž se vyvinují novou infekci během humorské léčby, zejména u pacientů, kteří dříve nebo nedávno cestovali do zemí s vysokou prevalencí tuberkulózy nebo kteří měli úzký kontakt s osobou s aktivní tuberkulózou.
Monitorování
Pečlivě sledujte pacienty ohledně rozvoje příznaků a příznaků infekce během a po léčbě humirou, včetně vývoje tuberkulózy u pacientů, kteří testovali negativní na latentní infekci tuberkulózy před zahájením terapie. Testy na infekci latentní tuberkulózy mohou být také nepravdivě negativní při terapii Humirou.
Přerušte humiru, pokud se pacient vyvine vážnou infekci nebo sepse. For a patient who develops a new infection during treatment with Humira closely monitor them perform a prompt a complete diagnostic workup appropriate for an immunocompromised patient a initiate appropriate antimicrobial therapy.
Invazivní plísňové infekce
Pokud se pacienti vyvinou vážnou systémovou onemocnění a pobývají nebo cestují v regionech, kde jsou mykosy endemické, zvažte invazivní plísňovou infekci v diferenciální diagnóze. U některých pacientů s aktivní infekcí může být testování antigenu a protilátek na histoplazmózu negativní. Zvažte vhodnou empirickou antimykotickou terapii s přihlédnutím k riziku závažné plísňové infekce a rizika antimykotiky, zatímco se provádí diagnostické zpracování. Abychom pomohli při léčbě těchto pacientů, zvažte konzultace s lékařem s odbornými znalostmi v oblasti diagnostiky a léčby invazivních plísňových infekcí.
Malignity
Zvažte rizika a přínosy léčby blokátorů TNF včetně Humira před zahájením terapie u pacientů se známou malignitou jinou než s úspěšně léčenou rakovinou nemelanomu (NMSC) nebo při zvažování pokračování blokátoru TNF u pacientů, kteří se vyvinou malignitu.
Malignity In Dospělí
V kontrolovaných částech klinických studií některých blokátorů TNF, včetně Humira, bylo pozorováno více případů malignit u dospělých pacientů ošetřených TNF-blokátorem ve srovnání s dospělými pacienty ošetřenými kontrolou. Během kontrolovaných částí 39 globálních humira klinických studií u dospělých pacientů s revmatoidní artritidou (RA) psoriatickou artritidou (PSA) ankylozující spondylitida (AS) Crohnovy choroby (CD) ulcerativní kolitida (UC) Plakes psoriasis (PS) a jiných psoriázů (UV) a jiných psoriasis (UV) (UV) (UV) (UV) (UV) a uV) a UV) a uV) a uV) a uV) a uV) psoriasitis (UV) a uV) a uV) a uV) a uV) a uV) a uV) a uV) a uV) a uV) a uV) a uV) a uV) a uV) a uV) a uV) UV) a uV) a uV) UV) a uV) Rakovina kůže nemelanomu (bazální buňka a skvamózní buňka) byla pozorována rychlostí (95% interval spolehlivosti) 0,7 (NULL,48 1,03) na 100 pacientových let u 7973 humira ošetřených pacientů ošetřených humirou (NULL,41 1,17) na 100 pacientů u 4848 pacientů s kontrolou pacientů a u pacientů s hladinou a u pacientů s hladinou) a u pacientů s tvorbou) a u pacientů s tvorbou) a u pacientů s tvorbou) a u pacientů s tvorbou) a u pacientů s hladinou) a zadržovali léčbu). V 52 globálních kontrolovaných a nekontrolovaných klinických studiích HUMIRA u dospělých pacientů s RA PSA jako CD UC PS HS a UV nejčastěji pozorovaných malignit jiných než lymfom a NMSC byly plicní prostaty a melanom prostaty prsu. Malignity u pacientů ošetřených humarou v kontrolovaných a nekontrolovaných částech studií byly podobné typu a počtu jako to, co by se dalo očekávat v obecné americké populaci podle databáze SEER (upraveno podle pohlaví a rasy) .1 .1.
V kontrolovaných studiích s jinými blokátory TNF u dospělých pacientů s vyšším rizikem malignit (tj. Pacienti s COPD s významnou anamnézou kouření a cyklofosfamidem léčeným pacienty s Wegenerovou granulomatózou) se ve skupině blokátorů TNF vyskytovala ve srovnání s kontrolní skupinou.
Rakovina kůže nemelanomu
Během kontrolovaných částí 39 globálních klinických studií HUMIRA u dospělých pacientů s RA PSA jako CD UC PS HS a UV rychlost (95% interval spolehlivosti) NMSC byla 0,8 (NULL,52 1,09) na 100 pacientů s pacientem u pacientů ošetřených HUMIRA a 0,2 (NULL,10 0,59) na 100 pacientů u pacientů s kontrolou. Prozkoumejte všechny pacienty a zejména pacienty s anamnézou předchozí prodloužené imunosupresivní terapie nebo pacienty s psoriázou s anamnézou léčby PUVA pro přítomnost NMSC před a během léčby Humirou.
Lymfom a leukémie
V kontrolovaných částech klinických studií všech blokátorů TNF u dospělých bylo pozorováno více případů lymfomu u pacientů ošetřených TNF-blokátorem ve srovnání s pacienty ošetřenými kontrolou. V kontrolovaných částech 39 globálních klinických studií Humira u dospělých pacientů s RA PSA jako CD UC PS HS a UV 2 lymfomy se vyskytly u 7973 pacientů ošetřených humirou oproti 1 u 4848 kontrolních pacientů. V 52 globálních kontrolovaných a nekontrolovaných klinických studiích HUMIRA u dospělých pacientů s RA PSA jako CD UC PS HS a UV s průměrnou dobou trvání přibližně 0,7 let včetně 24605 pacientů a více než 40215 pacientaears humira byla pozorovaná rychlost lymfomů přibližně 0,11 na 100 pacientů. To je přibližně 3krát vyšší, než se očekávalo v obecné americké populaci podle databáze SEER (upravená podle věku pohlaví a rasy) .1 Míra lymfomu v klinických studiích Humira nelze porovnat s mírou lymfomu v klinických studiích s jinými blokátory TNF a nemusí předpovídat míru pozorované v populaci pacientů. Pacienti s RA a jinými chronickými zánětlivými chorobami, zejména u pacientů s vysoce aktivním onemocněním a/nebo chronickou expozicí imunosupresivním terapiím, mohou být vystaveni vyššímu riziku (až do několikanásobků) než běžná populace pro rozvoj lymfomu i při absenci blokátorů TNF. Případy po trhu akutní a chronické leukémie byly hlášeny ve spojení s používáním blokátoru TNF v RA a dalšími indikacemi. Dokonce i při absenci pacientů s terapií blokátorů TNF mohou být vystaveny vyššímu riziku (přibližně 2krát) než běžná populace pro rozvoj leukémie.
Malignity In Pediatričtí pacienti And Young Dospělí
Malignity some fatal have been reported among children adolescents a young adults who received treatment with TNF-blockers (initiation of therapy ≤ 18 years of age) of which Humira is a member. Approximately half the cases were lymphomas including Hodgkin's a non-Hodgkin's lymphoma. The other cases represented a variety of different malignancies a included rare malignancies usually associated with immunosuppression a malignancies that are ne usually observed in children a adolescents. The malignancies occurčervený after a median of 30 months of therapy (range 1 to 84 months). Most of the patients were receiving concomitant immunosuppressants. These cases were reported post-marketing a are derived from a variety of sources including registries a spontaneous postmarketing reports.
U pacientů léčených blokátory TNF včetně HUMIRA byl hlášen ponorkové případy hepatosplenického lymfomu T-buněk (HSTCL) Vzácný typ lymfomu T-buněk. Tyto případy měly velmi agresivní průběh nemoci a byly fatální. Většina hlášených případů blokátoru TNF se vyskytla u pacientů s Crohnovou chorobou nebo ulcerózní kolitidou a většina byla u dospívajících a mladých dospělých mužů. Téměř všichni tito pacienti byli léčeni imunosupresivními azathioprin nebo 6-merkaptopurin (6-MP) současně s blokátorem TNF při nebo před diagnózou. Není jisté, zda výskyt HSTCL souvisí s použitím blokátoru TNF nebo blokátoru TNF v kombinaci s těmito dalšími imunosupresivami. Potenciální riziko s kombinací azathioprinu nebo 6mercaptopurinu a Humira by mělo být pečlivě zváženo.
Reakce přecitlivělosti
Po podání HUMIRA byly hlášeny anafylaxe a angioneurotický edém. Pokud dojde k anafylaktické nebo jiné závažné alergické reakci, okamžitě přeruší podávání Humira a Institute vhodné terapie. V klinických studiích s hypersenzitivními reakcemi Humira (např. Vyrážka anafylaktoidní reakce fixní léčiva, která není specifikovaná reakce léčiva).
Reaktivace viru viru hepatitidy B
Použití blokátorů TNF včetně Humira může zvýšit riziko reaktivace viru hepatitidy B (HBV) u pacientů, kteří jsou chronickými nosiči tohoto viru. V některých případech byla reaktivace HBV vyskytující se ve spojení s terapií blokátorů TNF fatální. Většina těchto zpráv se vyskytla u pacientů souběžně, kteří dostávali jiné léky, které potlačují imunitní systém, který může také přispět k reaktivaci HBV. Před zahájením terapie blokátory TNF hodnotí pacienty s rizikem infekce HBV pro předchozí důkaz infekce HBV. Pozornost při předepisování blokátorů TNF u pacientů identifikovaných jako nosiče HBV. Přiměřené údaje nejsou k dispozici o bezpečnosti nebo účinnosti léčby pacientů, kteří jsou nositeli HBV s antivirovou terapií ve spojení s terapií blokátorů TNF, aby se zabránilo reaktivaci HBV. U pacientů, kteří jsou nositeli HBV a vyžadují léčbu blokátorů TNF, pečlivě sledují takové pacienty, pokud jde o klinické a laboratorní příznaky aktivní infekce HBV během terapie a několik měsíců po ukončení terapie. U pacientů, u nichž se vyvinou reaktivaci HBV, zastavují humiru a zahájí účinnou antivirovou terapii při vhodné podpůrné léčbě. Bezpečnost obnovení terapie blokátorem TNF po kontrole reaktivace HBV není známa. Při zvažování obnovení humiry terapie v této situaci tedy dejte opatrnost a pečlivě sledujte pacienty.
Neurologické reakce
Použití blokovacích látek TNF včetně Humira bylo spojeno se vzácnými případy nového nástupu nebo exacerbace klinických symptomů a/nebo radiografického důkazu demyelinizačního onemocnění centrálního nervového systému včetně roztroušené sklerózy (MS) a optické neuritidy a periferní demyelinizační onemocnění včetně syndromu garrr. Pozornost při zvažování použití humiry u pacientů s předchozím nebo nedávným ústředním nebo periferním nervovým systémem demyelinizační poruchy; Pokud by se některá z těchto poruch vyvinula, mělo by se zvážit přerušení Humiry. Existuje známá asociace mezi mezilehlými uveitidami a centrálními demyelinizačními poruchami.
Hematologické reakce
U blokovacích látek TNF byly hlášeny vzácné zprávy o pancytopenii včetně aplastické anémie. Nežádoucí účinky hematologického systému včetně lékařsky významné cytopenie (např. Trombocytopenie leukopenie) byly s Humirou hlášeny zřídka. Příčinný vztah těchto zpráv k Humiru zůstává nejasný. Doporučujte všem pacientům, aby hledali okamžitou lékařskou péči, pokud si vyvinou příznaky a symptomy naznačující krevní dyscrasias nebo infekci (např. Přetrvávající pohmožděná horečka krvácející bledost) na Humiru. Zvažte přerušení terapie Humira u pacientů s potvrzenými významnými hematologickými abnormalitami.
Zvýšené riziko infekce při použití s anakinrou
Souběžné použití Anakinry (antagonisty interleukin-1) a dalšího blokátoru TNF bylo spojeno s větší podílem vážných infekcí a neutropenie a ve srovnání s blokátorem TNF u pacientů s RA bez dalšího přínosu. Proto se nedoporučuje kombinace Humira a Anakinra [viz Lékové interakce ].
Selhání srdce
U blokátorů TNF byly hlášeny případy zhoršujícího se městnavého srdečního selhání (CHF) a nového nástupu CHF. Případy zhoršujícího se CHF byly také pozorovány u Humira. Humira nebyla formálně studována u pacientů s CHF; V klinických studiích s jiným blokátorem TNF však byla pozorována vyšší míra závažných nežádoucích účinků souvisejících s CHF. Při používání Humiry u pacientů, kteří mají srdeční selhání, uchovávejte opatrnost a pečlivě je sledujte.
Autoimunita
Léčba Humirou může vést k vytvoření autoprotilátek a zřídka ve vývoji syndromu podobného lupusu. Pokud se pacient vyvine příznaky, které naznačují syndrom podobný lupusu po léčbě humirou, která přerušila léčbu [viz Nežádoucí účinky ].
Imunizace
V placebem kontrolované klinické studii pacientů s RA nebyla detekována v antipneumokokokové protilátkové odpovědi mezi humirou a placebem léčenou skupinami, když byla souběžně podána pneumokoková vakcína a vakcína proti chřipce s Humirou. Podobné proporce pacientů se vyvinuly ochranné úrovně anti-influenzových protilátek mezi skupinami ošetřování humira a placebem; Avšak titry v agregátu proti chřipce antigenů byly mírně nižší u pacientů, kteří dostávali humiru. Klinický význam tohoto není znám. Pacienti na Humiru mohou dostávat souběžné očkování s výjimkou živých vakcín. O sekundárním přenosu infekce nejsou k dispozici žádné údaje živými vakcínami u pacientů, kteří dostávají humiru.
Doporučuje se, aby byli pediatričtí pacienti, pokud je to možné, aktuální se všemi imunizacemi v souladu se současnými imunizačními pokyny před zahájením terapie humirou. Pacienti na Humiru mohou dostávat souběžné očkování s výjimkou živých vakcín.
Bezpečnost podávání živých nebo živých vakcín u kojenců vystavených Humiru v děloze je neznámé. Před očkováním (živé nebo živé nebo živé) exponované kojence by měla být zvážena rizika a výhody [viz viz Použití v konkrétních populacích ].
Zvýšené riziko infekce při použití u abataceptu
V kontrolovaných studiích bylo souběžné podávání blokátorů TNF a abataceptu spojeno s větší podílem vážných infekcí než použití samotného blokátoru TNF; Kombinovaná terapie ve srovnání s používáním samotného blokátoru TNF neprokázala zlepšený klinický přínos při léčbě RA. Proto se nedoporučuje kombinace abataceptu s blokátory TNF včetně HUMIRA [viz viz Lékové interakce ].
Informace o poradenství pro pacienta
Poraďte se s pacientem nebo pečovatelem, aby si přečetl značení pacienta schváleného FDA ( Průvodce léky a Pokyny pro použití ).
Infekce
Informujte pacienty, že Humira může snížit schopnost jejich imunitního systému bojovat proti infekcím. Instrujte pacienty o důležitosti kontaktování svého lékaře, pokud si vyvinou jakékoli příznaky infekce, včetně invazivních plísňových infekcí tuberkulózy a reaktivace infekcí viru hepatitidy B [viz viz VAROVÁNÍS AND OPATŘENÍ ].
Malignity
Poraden pacientům o riziku malignit při přijímání humiry [viz VAROVÁNÍS AND OPATŘENÍ ]
Reakce přecitlivělosti
Poraďte pacientům, aby hledali okamžitou lékařskou péči, pokud zažívají jakékoli příznaky závažných hypersenzitivních reakcí. Poraďte se s pacienty citlivými na latex, že jehla Humira 40 mg/0,8 ml pera a 40 mg/0,8 ml 20 mg/0,4 ml a 10 mg/0,2 ml předběžné stříkačky může obsahovat latex přirozené gumy [viz viz VAROVÁNÍS AND OPATŘENÍ Jak dodáno / Skladování a manipulace ].
Ostatní Medical Conditions
Poraďte pacientům, aby hlásili jakékoli známky nového nebo zhoršujícího se zdravotního stavu, jako jsou kongestivní srdeční selhání neurologické onemocnění autoimunitních poruch nebo cytopenie. Poraďte pacientům, aby nahlásili jakékoli příznaky naznačující cytopenii, jako je krvácení z modřin nebo přetrvávající horečka [viz VAROVÁNÍS AND OPATŘENÍ ].
Pokyny k technice injekce
Informujte pacienty, že první injekce má být prováděna pod dohledem kvalifikovaného zdravotnického pracovníka. Má -li je pacient nebo pečovatel podávat HUMIRA, instrukci v injekčních technikách a posoudit jejich schopnost vstřikovat subkutánně, aby zajistil správné podávání Humiry [viz viz Pokyny pro použití ].
Pro pacienty, kteří budou používat HUMIRA PEN, jim řeknou, že:
- Když je stisknuto tlačítko aktivátoru plum-zbarveného aktivace, uslyší hlasitý kliknutí. Hlasité kliknutí znamená začátek injekce.
- Musí držet HUMIRA pero proti jejich vymaččené zvednuté kůži, dokud se vstříkne veškerý lék. To může trvat až 15 sekund.
- Bude vědět, že injekce skončila, když se žlutá značka plně objeví v pohledu okna a přestane se pohybovat.
Poskytněte pacientům, aby zlikvidovali použité jehly a injekční stříkačky nebo použily pero v nádobě na likvidaci Sharps čištěné FDA bezprostředně po použití. Poskytněte pacientům, aby do jejich domácnosti nezakládali volné jehly a injekční stříkačky nebo pero. Poskytněte pacientům, že pokud nemají nádobu na likvidaci Sharps, které nemají FDA, mohou používat kontejner pro domácnost, která je vyrobena z těžkých plastů, může být uzavřena pevně přiléhajícím a propíchnutím odolným víkem, aniž by ostře, aniž by byly schopny vyjít vzpřímeně a stabilní během používání odolných proti úniku a náležitě označené k válce s rizikovým odpadem.
Poskytněte pacientům, že když je jejich nádoba na likvidaci SHARPS téměř plná, budou muset dodržovat své komunitní pokyny pro správný způsob, jak zlikvidovat jejich nádobu na likvidaci ostře. Poskytněte pacientům, že mohou existovat státní nebo místní zákony týkající se likvidace použitých jehel a stříkaček. Další informace o bezpečném likvidaci Sharps a konkrétních informacích o likvidaci Sharps ve stavu, ve kterém žijí, naleznete pacienty na webu FDA na adrese https://www.fda.gov/safesharpsdisposal.
Poskytněte pacientům, aby nekvalifikovali svůj použitý kontejner s ostře na likvidaci ve svém odpadku pro domácnost, pokud to jejich pokyny pro komunitu neumožňují. Poskytněte pacientům, aby nerecyktovali jejich použitý nádobu na likvidaci Sharps.
Neklinická toxikologie
Zhodnocení mutageneze karcinogeneze plodnosti
Dlouhodobé studie na zvířatech Humira nebyly provedeny za účelem vyhodnocení karcinogenního potenciálu nebo jeho účinku na plodnost.
Použití v konkrétních populacích
Těhotenství
Shrnutí rizika
Dostupné studie s použitím adalimumabu během těhotenství spolehlivě nestanují souvislost mezi adalimumabem a hlavními vrozenými vadami. Klinické údaje jsou k dispozici od Organizace teratologických informačních specialistů (OTIS)/Mothertobaby Humira těhotenství u těhotných žen s revmatoidní artritidou (RA) nebo Crohnovou chorobou (CD). Výsledky registru ukázaly míru 10% u hlavních vrozených vad s použitím Adalimumabu prvního trimestru u těhotných žen s RA nebo CD a míra 7,5% u hlavních vrozených vad v nepokojné srovnávací kohortě. Nedostatek vzoru hlavních vrozených vad je uklidňující a rozdíly mezi expozičními skupinami mohly ovlivnit výskyt vrozených vad (viz viz Data ).
Adalimumab je aktivně přenášen napříč placentou během třetího trimestru těhotenství a může ovlivnit imunitní reakci u unero exponovaného dítěte (viz Klinické úvahy ). In an embryo-fetal perinatal development study conducted in cynomolgus monkeys no fetal harm or malformations were observed with intravenous administration of Adalimumab during organogenesis a later in gestation at doses that produced exposures up to approximately 373 times the maximum recommended human dose (MRHD) of 40 mg subcutaneous without methotrexate (see Data ).
Odhadované riziko pozadí hlavních vrozených vad a potratu pro uvedené populace není známo. Všechna těhotenství mají na pozadí riziko ztráty vrozených vad nebo jiných nepříznivých výsledků. V americké obecné populaci je odhadované riziko na pozadí hlavních vrozených vad a potratu u klinicky uznávaných těhotenství 2-4% a 15–20%.
Klinické úvahy
Mateřské a embryo/fetální riziko spojené s nemocí
Zveřejněné údaje naznačují, že riziko nepříznivých výsledků těhotenství u žen s RA nebo zánětlivé onemocnění střev (IBD) je spojeno se zvýšenou aktivitou onemocnění. Nepříznivé výsledky těhotenství zahrnují předčasné porod (před 37 týdny těhotenství) nízká porodní hmotnost (méně než 2500 g) kojenců a malé pro gestační věk při narození.
Fetální/novorozenecké nežádoucí účinky
Monoklonální protilátky jsou stále více transportovány přes placentu, protože těhotenství postupuje s největším množstvím přeneseným během třetího trimestru (viz viz Data ). Risks a benefits should be considečervený prior to administering live or live-attenuated vaccines to infants exposed to Humira v děloze [see Použití v konkrétních populacích ].
Data
Lidská data
Registr expozice prospektivní kohortové těhotenství provedený společností Otis/Mothertobaby v USA a Kanadě mezi lety 2004 a 2016 porovnával riziko velkých vrozených vad u kojenců 221 žen (69 RA 152 CD) léčených adalimumabem během prvního trimestru a 106 žen (74 RA 32 CD), které nebylo léčeno adalimumabem.
Podíl hlavních vrozených vad mezi živě narozenými kojenci v adalimumabu ošetřených a neošetřených kohortách byl 10% (NULL,7% RA 10,5% CD) a 7,5% (NULL,8% RA 9,4% CD). Nedostatek vzoru hlavních vrozených vad je uklidňující a rozdíly mezi expozičními skupinami mohou ovlivnit výskyt vrozených vad. Tato studie nemůže spolehlivě zjistit, zda existuje souvislost mezi adalimumabem a hlavními vrozenými vadami kvůli metodickým omezením registru, včetně malé velikosti vzorku, dobrovolné povahy studie a ne-randomizovaného designu.
V nezávislé klinické studii prováděné u deseti těhotných žen s IBD léčeným koncentracemi Humira Adalimumab byly měřeny v mateřském séru i v pupečníkové krvi (n = 10) a kojeneckém séru (n = 8) v den narození. Poslední dávka Humira byla podávána mezi 1 a 56 dny před doručením. Koncentrace adalimumabu byly 0,16-19,7 μg/ml v pupečníkové krvi 4,28-17,7 μg/ml v kojeneckém séru a 0-16,1 μg/ml v mateřském séru. Ve všech případech kromě jednoho případu byla koncentrace adalimumabu s pupečníkem vyšší než koncentrace mateřského séra naznačující, že adalimumab aktivně protíná placentu. Navíc jedno dítě mělo koncentrace v séru v každém z následujících: 6 týdnů (NULL,94 μg/ml) 7 týdnů (NULL,31 μg/ml) 8 týdnů (NULL,93 μg/ml) a 11 týdnů (NULL,53 μg/ml), což může být detekováno v séru inféntů vystavených v utero od narození.
Údaje o zvířatech
V studii perinatálního vývoje v embryo-fetálních perinatálních vývojových studiích obdrželi opice cynomolgus opice adalimumab od dnů těhotenství 20 až 97 v dávkách, které produkovaly expozice až 373krát, které dosáhly s MRHD bez methotrexátu (na základě AUC s dávkami IV do 100 mg/kg/týdne). Adalimumab nevyvolával újmu na plody nebo malformace.
Laktace
Shrnutí rizika
Omezené údaje z případových zpráv v publikované literatuře popisují přítomnost adalimumabu v lidském mléce při dávkách kojenců 0,1% až 1% koncentrace mateřského séra. Zveřejněná data naznačují, že se očekává, že systémová expozice kojenému dítěti bude nízká, protože adalimumab je velká molekula a je degradována v gastrointestinálním traktu. Účinky lokální expozice v gastrointestinálním traktu však nejsou známy. Neexistují žádné zprávy o nepříznivých účincích adalimumabu na kojené dítě a žádné účinky na produkci mléka. Vývojové a zdravotní přínosy kojení by měly být zváženy spolu s klinickou potřebou matky pro humiru a jakékoli potenciální nepříznivé účinky na kojené dítě z humiry nebo ze základního mateřského stavu.
Dětské použití
Bezpečnost a účinnost HUMIRA byla stanovena pro:
- Snížení příznaků a příznaků mírně na těžce aktivní polyartikulární JIA u pediatrických pacientů ve věku 2 let a starší.
- Léčba mírně až vážně aktivní Crohnovy choroby u pediatrických pacientů ve věku 6 let a starších.
- Léčba mírně až vážně aktivní ulcerativní kolitidy u pediatrických pacientů ve věku 5 let a starších.
- Léčba střední až těžká hidradenitida Supsurativa u pacientů ve věku 12 let a starších.
- Léčba neinfekční střední a panuveitidy u pediatrických pacientů ve věku 2 let a starších.
Vzhledem k jeho inhibici TNFa Humira podávané během těhotenství by mohla ovlivnit imunitní odpověď v novorozeneckém a kojenci v utero-exponovaném. Údaje od osmi kojenců vystavených Humiru v děloze naznačují, že Adalimumab prochází placenta [viz Použití v konkrétních populacích ]. The clinical significance of elevated Adalimumab concentrations in infants is unknown. The safety of administering live or live-attenuated vaccines in exposed infants is unknown. Risks a benefits should be considečervený prior to vaccinating (live or live-attenuated) exposed infants.
Případy lymfomu po trhu včetně hepatosplenického lymfomu T-buněk a dalších malignit byly hlášeny některé fatální mezi dětmi adolescenty a mladými dospělými, kteří dostávali léčbu blokátorům TNF včetně HUMIRA [viz viz HUMIRA [viz viz HUMIRA [viz viz HUMIRA [viz viz HUMIRA [viz viz HUMIRA [viz viz Humira [viz HUMIRA [viz VAROVÁNÍS AND OPATŘENÍ ].
Juvenilní idiopatická artritida
Ve studii bylo prokázáno, že Jia-I Humira snižuje příznaky a příznaky aktivní polyartikulární JIA u pacientů 4 až 17 let [viz Klinické studie ]. In Studujte jia-iI the safety profile for patients 2 to <4 years of age was similar to the safety profile for patients 4 až 17 let with polyarticular JIA [see Nežádoucí účinky ]. Humira has ne been studied in patients with polyarticular JIA less than 2 years of age or in patients with a weight below 10 kg.
Bezpečnost humira u pacientů v polyartikulárních studiích JIA byla obecně podobná bezpečnosti pozorované u dospělých s určitými výjimkami [Viz Nežádoucí účinky ].
U pediatrických pacientů s JIA nebyla stanovena bezpečnost a účinnost Humiry.
Pediatrická Crohnova nemoc
U pediatrických pacientů ve věku a starších a starších a starších věku a starších nemocí byla stanovena bezpečnost a účinnost HUMIRA pro mírně aktivní Crohnovou chorobu. Použití humir pro tuto indikaci je podporováno důkazy z přiměřených a dobře kontrolovaných studií u dospělých s dodatečnými údaji z randomizované dvojitě slepé 52týdenní klinické studie dvou koncentrací dávky v roce 192 pediatrických pacientů (6 let až 17 let) [Viz) Nežádoucí účinky Klinická farmakologie Klinické studie ]. The adverse reaction profile in patients 6 years to 17 years of age was similar to adults.
U pediatrických pacientů s Crohnovou chorobou mladší 6 let nebyla stanovena bezpečnost a účinnost Humiry.
Pediatrická ulcerózní kolitida
U pediatrických pacientů a starších a starších a starších byla stanovena bezpečnost a účinnost Humiry pro léčbu mírně až vážně aktivní ulcerózní kolitidy. Použití humir pro tuto indikaci je podporováno důkazy z přiměřených a dobře kontrolovaných studií u dospělých s dalšími údaji z randomizované dvojitě zaslepené 52týdenní klinické studie dvou koncentrací dávky Humira u 93 dětských pacientů (5 let až 17 let) [Viz) Nežádoucí účinky Klinická farmakologie Klinické studie ]. The adverse reaction profile in patients 5 years to 17 years of age was similar to adults.
Účinnost Humiry nebyla stanovena u pacientů, kteří ztratili odpověď nebo byli netolerantní na blokátory TNF.
U pediatrických pacientů s ulcerativní kolitidou mladší než 5 let nebyla stanovena bezpečnost a účinnost Humiry.
Dětská uveitida
U pediatrických pacientů a starších věku a starších byla stanovena bezpečnost a účinnost Humiry pro léčbu neinfekční uveitidy. Použití Humiry je podporováno důkazy z přiměřených a dobře kontrolovaných studií humir u dospělých a náhodnou kontrolovanou klinickou studií 2: 1 u 90 pediatrických pacientů [viz viz Klinické studie ]. The safety a effectiveness of Humira have ne been established in pediatric patients with uveitis less than 2 years of age.
Hidradenitis Supsurative
Použití humira u pediatrických pacientů ve věku 12 let a starších pro HS je podporováno důkazy z přiměřených a dobře kontrolovaných studií humir u dospělých pacientů s HS. Další populační farmakokinetické modelování a simulace předpovídala, že dávkování Humiry založené na hmotnosti u dětských pacientů ve věku 12 let a starší může obecně podobné expozici dospělým pacientům s HS. Průběh HS je u dospělých a dospívajících pacientů dostatečně podobný, aby umožňoval extrapolaci údajů od dospělých na dospívající pacienty. Doporučená dávka u dětských pacientů ve věku 12 let nebo starší je založena na tělesné hmotnosti [viz Dávkování a podávání Klinická farmakologie a Klinické studie ].
Bezpečnost a účinnost Humiry nebyla u pacientů mladších 12 let stanovena s HS.
Geriatrické použití
Celkem 519 pacientů s RA ve věku 65 let a starších včetně 107 pacientů ve věku 75 let a starších dostávalo Humiru v klinických studiích RA-I až IV. Mezi těmito pacienty a mladšími pacienty nebyl pozorován žádný celkový rozdíl v účinnosti. Frekvence vážné infekce a malignity u pacientů s humirou ve věku 65 let a starších byla vyšší než u pacientů mladších 65 let. Zvažte výhody a rizika Humira u pacientů ve věku 65 let a starší. U pacientů léčených Humirou pečlivě sleduje vývoj infekce nebo malignity [viz VAROVÁNÍS AND OPATŘENÍ ].
Informace o předávkování pro Humiru
Dávky až 10 mg/kg byly podávány pacientům v klinických studiích bez důkazu toxicity omezující dávku. V případě předávání se doporučuje, aby byl pacient sledován na jakékoli příznaky nebo příznaky nežádoucích účinků nebo účinků a vhodné symptomatické léčby bylo okamžitě zavedeno.
Kontraindikace pro Humiru
Žádný.
Klinická farmakologie for Humira
Mechanismus působení
Adalimumab se váže konkrétně na TNF-alfa a blokuje svou interakci s TNF receptory buněčného povrchu p55 a p75. Adalimumab také lyses povrch TNF exprimující buňky in vitro v přítomnosti komplementu. Adalimumab se neváže ani neaktibuje lymfotoxin (TNF-beta). TNF je přirozeně se vyskytující cytokin, který se podílí na normálních zánětlivých a imunitních odpovědích. Zvýšené koncentrace TNF se nacházejí v synoviální tekutině u pacientů s RA jia PSA a AS a hrají důležitou roli jak v patologickém zánětu, tak v destrukci kloubů, které jsou charakteristickými znaky těchto onemocnění. Zvýšené koncentrace TNF se také nacházejí v plaketách psoriázy. Při léčbě PS Humirou může snížit epidermální tloušťku a infiltraci zánětlivých buněk. Vztah mezi těmito farmakodynamickými aktivitami a mechanismem (mechanismy), kterými Humira vyvíjí své klinické účinky, není znám.
Adalimumab také moduluje biologické reakce, které jsou indukovány nebo regulovány TNF, včetně změn v koncentracích adhezních molekul odpovědných za migraci leukocytů (ELAM-1 VCAM-1 a ICAM-1 s IC50 1-2 x 10-10M).
Farmakodynamika
Po léčbě Humirou bylo pozorováno snížení koncentrací reaktantů akutní fáze zánětu (C-reaktivní protein [CRP] a rychlost sedimentace erytrocytů [ESR]) a sérových cytokinů (IL-6) ve srovnání s výchozím hodnotou u pacientů s reumatoidní artritidou. Snížení koncentrací CRP bylo také pozorováno u pacientů s Crohnovou onemocněním ulcerativní kolitidou a hidradenitidou Supsurativa. Sérové koncentrace matricových metaloproteináz (MMP-1 a MMP-3), které způsobují remodelaci tkáně zodpovědné za destrukci chrupavky, byly také sníženy po podání humiry.
U pediatrických pacientů 5 let až 17 let s ulcerativní kolitidou je doporučená dávka Humira založena na modelovaných vztazích s dávkou/expozicí a efektivitou a farmakokinetická data. Neočekávané klinicky relevantní rozdíly v účinnosti mezi studovaným vyšší dávkou podávanou v klinické studii (týdny 0 až 52 ve studii puc-i) [Viz viz Klinické studie ] a doporučené dávkování [viz Dávkování a podávání ].
Farmakokinetika
Farmakokinetika Adalimumabu byla lineární v rozmezí dávky 0,5 až 10 mg/kg po podání jediné intravenózní dávky (Humira není schválena pro intravenózní použití). Po 20 40 a 80 mg každý druhý týden a každý týden subkutánní podávání adalimumab střední koncentrace séra v ustáleném stavu se zvýšila přibližně úměrně s dávkou u pacientů s RA. Průměrný terminální poločas byl přibližně 2 týdny v rozmezí od 10 do 20 dnů napříč studiemi. Zdravé subjekty a pacienti s RA vykazovali podobnou adalimumab farmakokinetiku.
Odhaduje se, že expozice adalimumabu u pacientů léčených 80 mg každý druhý týden je srovnatelná u pacientů léčených 40 mg každý týden.
Vstřebávání
Průměrná absolutní biologická dostupnost adalimumabu po jedné podkožní dávce 40 mg byla 64%. Průměrná doba k dosažení maximální koncentrace byla 5,5 dne (131 ± 56 hodin) a maximální koncentrace v séru byla 4,7 ± 1,6 mcg/ml u zdravých subjektů po jediném 40 mg subkutánním podávání humira.
Rozdělení
Distribuční objem (VSS) se pohyboval od 4,7 do 6,0 l po intravenózním podávání dávek v rozmezí od 0,25 do 10 mg/kg u pacientů s RA.
Odstranění
Jednorázová farmakokinetika adalimumabu u pacientů s RA byla stanovena v několika studiích s intravenózními dávkami v rozmezí od 0,25 do 10 mg/kg. Systémová clearance adalimumabu je přibližně 12 ml/h. V dlouhodobých studiích s dávkováním déle než dva roky nedošlo u pacientů s RA v průběhu času žádné důkazy o změnách clearance.
Populace pacientů
Revmatoidní artritida And Ankylozující spondylitida
U pacientů, kteří dostávali 40 mg humira každý druhý týden, byly průměrné koncentrace koryta v ustáleném stavu přibližně 5 mcg/ml a 8 až 9 mcg/ml bez as MTX souběžné léčby. Koncentrace adalimumabu v synoviální tekutině od pěti pacientů s revmatoidní artritidou se pohybovaly od 31 do 96% pacientů v séru. Farmakokinetika adalimumabu u pacientů s AS byla podobná u pacientů s RA.
Psoriatická artritida
U pacientů, kteří dostávali 40 mg každý druhý týden, byly průměrné koncentrace koryta v ustáleném stavu 6 až 10 mcg/ml a 8,5 až 12 mcg/ml bez as MTX souběžné léčby.
Plakesová psoriáza
Průměrná koncentrace koryta v ustáleném stavu byla přibližně 5 až 6 mcg/ml během ošetření Humira 40 mg každý druhý týden.
Dospělý uveitida
Adalimumab průměrná stabilní koncentrace byla přibližně 8 až 10 mcg/ml během HUMIRA 40 mg každý druhý týden.
Dospělý Hidradenitis Supsurative
Koncentrace koryta Adalimumab byly přibližně 7 až 8 mcg/ml ve 2. týdnu a 4, respektive týden po obdržení 160 mg v týdnu 0, po které následovalo 80 mg na 2. týden. Průměrné koncentrace koryta v ustáleném stavu ve 12. až 36. týdnu byly přibližně 7 až 11 mcg/ml během zacházení s humirou 40 mg.
Nemoc dospělého Crohna
Průměrné koncentrace koryta Adalimumab byly přibližně 12 mcg/ml ve 2. a 4. týdnu po obdržení 160 mg v týdnu 0, po které následovalo 80 mg na 2. týden. Průměrné koncentrace koryta v ustáleném stavu byly 7 mcg/ml v týdnu 24 a týden 56 během léčby HUMIRA 40 mg.
Ulcerativní kolitida dospělých
Průměrné koncentrace koryta Adalimumab byly přibližně 12 mcg/ml ve 2. a 4. týdnu po obdržení 160 mg v týdnu 0, po kterém následovalo 80 mg na 2. týden. Průměrné koncentrace koryta v ustáleném stavu byly přibližně 8 mcg/ml a 15 mcg/ml v týdnu po 52. týdnu po 52. týdnu po dobu 52 týdnů.
Účinky protilátky proti léčivo na farmakokinetiku
Revmatoidní artritida
Byl identifikován trend směrem k vyšší zjevné clearance adalimumabu v přítomnosti protilátek anti-adalimumab.
Pediatrická ulcerózní kolitida
Protilátky proti adalimumabu pomocí testu ECL byly spojeny se sníženými koncentracemi adalimumabu v séru u pediatrických pacientů s mírně až vážně aktivní ulcerativní kolitidou.
Hidradenitis Supsurative
U subjektů se středně těžkým až závažným protilátkami HS proti adalimumabu byla spojena se sníženými koncentracemi adalimumabu v séru. Obecně je rozsah snížení koncentrací séra adalimumab větší se zvyšující se titry protilátek proti adalimumabu.
Konkrétní populace
Geriatričtí pacienti
U pacientů s RA ve věku 40 až 75 let byla pozorována nižší vůle s rostoucím věkem.
Pediatričtí pacienti
Juvenilní idiopatická artritida
- 4 roky až 17 let: průměrné koncentrace koryta v ustáleném stavu byly 6,8 mcg/ml a 10,9 mcg/ml u pacientů vážících pacienty <30 kg receiving 20 mg Humira subcutaneously every other week as moneherapy or with concomitant MTX respectively. The Adalimumab mean steady-state trough concentrations were 6.6 mcg/mL a 8.1 mcg/mL in patients weighing ≥30 kg receiving 40 mg Humira subcutaneously every other week as moneherapy or with MTX concomitant treatment respectively.
- 2 roky do <4 years of age or 4 years of age a older weighing <15 kg: The Adalimumab mean steady-state trough Adalimumab concentrations were 6.0 mcg/mL a 7.9 mcg/mL in patients receiving Humira subcutaneously every other week as moneherapy or with MTX concomitant treatment respectively.
Pediatrická hidradenitida Supsurativa
Předpokládá se, že koncentrace adalimumabu u adolescentních pacientů s HS dostávajícími doporučené dávkové režimy jsou podobné koncentracím pozorovaným u dospělých subjektů s HS na základě farmakokinetického modelování populace a simulaci.
Pediatrická Crohnova choroba
Adalimumab průměrné koncentrace ± SD byly 15,7 ± 6,5 mcg/ml ve 4. týdnu po 160 mg v týdnu 0 a 80 mg v týdnu 2 a 10,5 ± 6,0 mcg/ml v týdnu 52 po 40 mg každý druhý týden u pacientů vážících váhu vážící ≥ 40 kg. Adalimumab průměrné koncentrace ± SD byly 10,6 ± 6,1 mcg/ml ve 4. týdnu po dávkování 80 mg v týdnu 0 a 40 mg v týdnu 2 a 6,9 ± 3,6 mcg/ml v týdnu 52 po 20 mg po 20 mg každý druhý týden u pacientů s vážením <40 kg.
Pediatrická ulcerózní kolitida
Průměrná koncentrace koryta v ustáleném stavu adalimumabu byla 52 mcg/ml v 52. týdnu po subkutánním podání 0,6 mg/kg (maximálně 40 mg) každý druhý týden u pediatrických pacientů s UC 5 let až 17 let. U pacientů, kteří dostávali 0,6 mg/kg (maximálně 40 mg) každý týden, byla průměrná koncentrace koryta v ustáleném stavu 15,7 ± 5,6 mcg/ml v týdnu 52 u pediatrických pacientů s UC 5 let do 17 let.
Pacienti s muži a ženy
Po korekci pro tělesnou hmotnost pacienta nebyly pozorovány žádné farmakokinetické rozdíly související s pohlavími. Zdravé subjekty a pacienti s revmatoidní artritidou vykazovali podobnou adalimumab farmakokinetiku.
Pacienti s renálním nebo jaterním poškozením
U pacientů s jaterním nebo renálním poškozením nejsou k dispozici žádné farmakokinetické údaje.
Revmatoidní faktor nebo koncentrace CRP
U pacientů s RA, kteří dostávali dávky nižší než doporučená dávka a u pacientů s vysokým revmatoidním faktorem nebo koncentrací CRP, bylo předpovídáno drobné zvýšení zjevné clearance. Tato zvýšení pravděpodobně nebude klinicky důležitá.
Studie interakce léčiva
Methotrexát
MTX snížil adalimumab zjevnou clearance po jednom a vícenásobném dávkování o 29% a 44% u pacientů s RA [viz viz Lékové interakce ].
Klinické studie
Revmatoidní artritida
Účinnost a bezpečnost humir byla hodnocena v pěti randomizovaných dvojitě slepých studiích u pacientů ve věku ≥18 let s aktivní revmatoidní artritidou (RA) diagnostikovanou podle kritérií American College of Reumatology (ACR). Pacienti měli nejméně 6 oteklých a 9 jemných kloubů. Humira byl podáván subkutánně v kombinaci s methotrexátem (MTX) (NULL,5 až 25 mg studií RA-I RA-III a RA-V) nebo jako monoterapie (studie RA-II a RA-V) nebo s jinými anti-rheumatickými léky (DMARDS) (Studie RA-IV).
Studie RA-I hodnotila 271 pacientů, kteří selhali terapií alespoň jednou, ale ne více než čtyř DMARD a neměli nedostatečnou reakci na MTX. Dávky 20 40 nebo 80 mg Humiry nebo placeba byly podávány každý druhý týden po dobu 24 týdnů.
Studie RA-II hodnotila 544 pacientů, kteří selhali terapií alespoň s jedním DMARD. Dávky placeba 20 nebo 40 mg humira byly podávány jako monoterapie každý druhý týden nebo týdně po dobu 26 týdnů.
Studie RA-III hodnotila 619 pacientů, kteří měli nedostatečnou reakci na MTX. Pacienti dostávali placebo 40 mg Humira každý druhý týden s injekcemi placeba v alternativních týdnech nebo 20 mg Humira týdně po dobu 52 týdnů. Studie RA-III měla další primární koncový bod po 52 týdnech inhibice progrese onemocnění (jak bylo zjištěno podle rentgenových výsledků). Po dokončení prvních 52 týdnů 457 pacientů zapsaných do fáze prodloužení s otevřenou značkou, ve které bylo 40 mg Humiry podáno každý druhý týden po dobu až 5 let.
Studie RA-IV vyhodnotila bezpečnost u 636 pacientů, kteří byli buď DMARD-NAIVE nebo bylo povoleno zůstat na své již existující revmatologické terapii za předpokladu, že terapie byla stabilní po dobu minimálně 28 dnů. Pacienti byli randomizováni na 40 mg Humiru nebo placeba každý druhý týden po dobu 24 týdnů.
Studie RA-V vyhodnotila 799 pacientů s mírně až vážně aktivní RA kratší než 3 roky, kteří byli ve věku ≥ 18 let a MTX Naãve. Pacienti byli randomizováni, aby dostávali buď MTX (optimalizovaný na 20 mg/týden do 8. týdne) Humira 40 mg každý druhý týden, nebo kombinovaná terapie Humira/MTX po dobu 104 týdnů. Pacienti byli hodnoceni na příznaky a symptomy a na radiografický progresi poškození kloubů. Střední doba trvání onemocnění u pacientů zapsaných do studie byla 5 měsíců. Střední dosažená dávka MTX byla 20 mg.
Klinická odpověď
Procento pacientů s ošetřením Humira dosahujících ACR 20 50 a 70 odpovědí ve studiích RAII a III je uvedeno v tabulce 3.
Tabulka 3: Odpovědi ACR ve studiích RA-II a RA-III (procento pacientů)
| Odpověď | Studium monoterapie RA-II (26 týdnů) | Studujte kombinaci methotrexátu RA-III (24 a 52 týdnů) | |||
| Placebo N = 110 | Humira 40 mg každý druhý týden N = 113 | Humira 40 mg weekly N = 103 | Placebo/ MTX N = 200 | Humira/ MTX 40 mg každý druhý týden N = 207 | |
| ACR20 | |||||
| 6. měsíc | 19% | 46%* | 53%* | 30% | 63%* |
| 12. měsíc | Na | Na | Na | 24% | 59%* |
| ACR50 | |||||
| 6. měsíc | 8% | 22%* | 35%* | 10% | 39%* |
| 12. měsíc | Na | Na | Na | 10% | 42%* |
| ACR70 | |||||
| 6. měsíc | 2% | 12%* | 18%* | 3% | 21%* |
| 12. měsíc | Na | Na | Na | 5% | 23%* |
| * P. <0.01 Humira vs. placebo |
Výsledky studie RA-I byly podobné studii RA-III; Pacienti, kteří dostávali Humiru 40 mg každý druhý týden ve studii RA-I, také dosáhli míry odezvy ACR 20 50 a 70 65% 52% a 24% ve srovnání s placebem 13% 7% a 3% po 6 měsících (P <0.01).
Výsledky složek kritéria odezvy ACR pro studie RA-II a RA-III jsou uvedeny v tabulce 4. Míra odezvy ACR a zlepšení ve všech složkách ACR reakce bylo udržováno na 104. týdne. Během 2 let ve studii Ra-III 20% HUMIRA pacientů, kteří dostávali 40 mg, každý další týden dosáhl hlavních klinických reakcí na 6-měsíční období. Reakce ACR byly udržovány v podobných poměrech pacientů po dobu až 5 let s kontinuální léčbou humira v otevřené části studie RA-III.
Tabulka 4: Složky odpovědi ACR ve studiích RA-II a RA-III
| Parametr (medián) | Studujte ra-ii | Studujte ra-iiI | ||||||
| Placebo N = 110 | Humira a N = 113 | Placebo/Mtx N = 200 | Humira a /Mtx N = 207 | |||||
| Základní linie | Wk 26 | Základní linie | Wk 26 | Základní linie | WK 24 | Základní linie | WK 24 | |
| Počet jemných kloubů (0-68) | 35 | 26 | 31 | 16* | 26 | 15 | 24 | 8* |
| Počet oteklých kloubů (0-66) | 19 | 16 | 18 | 10* | 17 | 11 | 18 | 5* |
| Globální hodnocení lékaře b | 7.0 | 6.1 | 6.6 | 3.7* | 6.3 | 3.5 | 6.5 | 2,0* |
| Globální hodnocení pacienta b | 7.5 | 6.3 | 7.5 | 4,5* | 5.4 | 3.9 | 5.2 | 2,0* |
| Bolest b | 7.3 | 6.1 | 7.3 | 4.1* | 6.0 | 3.8 | 5.8 | 2.1* |
| Index postižení (HAQ) c | 2.0 | 1.9 | 1.9 | 1,5* | 1.5 | 1.3 | 1.5 | 0,8* |
| CRP (MG/DL) | 3.9 | 4.3 | 4.6 | 1,8* | 1.0 | 0.9 | 1.0 | 0,4* |
| a 40 mg Humira se podává každý druhý týden b Vizuální analogová stupnice; 0 = nejlepší 10 = nejhorší c Index zdravotního postižení dotazníku pro hodnocení zdraví; 0 = Nejlepší 3 = nejhorší měření schopnost pacienta provádět následující: Dress/Grise Eat Eat Walk Reach Grip Hygiene a udržovat každodenní aktivitu * P. <0.001 Humira vs. placebo based on mean change from baseline |
Časový průběh reakce ACR 20 pro studii RA-III je znázorněn na obrázku 1.
Ve studii RA-III 85% pacientů s odpověďmi ACR 20 ve 24. týdnu udržovalo reakci po 52 týdnech. Časový průběh reakce ACR 20 pro studii RA-I a studium RA-II byl podobný.
Obrázek 1: Studujte RA-III ACR 20 Reakce za 52 týdnů
 |
Ve studii RA-IV 53% pacientů léčených Humirou 40 mg každý druhý týden plus standard péče měl odpověď ACR 20 ve 24. týdnu ve srovnání s 35% na placebu plus standardní péči (P <0.001). No unique adverse reactions related to the combination of Humira (Adalimumab) a other DMARDs were observed.
Ve studii RA-V s pacienty s MTX Na¯ve s nedávným nástupem RA vedla kombinovaná léčba s Humirou plus MTX k většímu procentu pacientů s reakcemi na ACR než monoterapie MTX nebo monoterapii HUMIRA v 52. týdnu a odpovědi byly udržovány v týdnu 104 (viz tabulka 5).
Tabulka 5: Reakce ACR ve studii RA-V (procento pacientů)
| Odpověď | MTX b N = 257 | Humira c N = 274 | Humira/Mtx N = 268 |
| ACR20 | |||
| 52 týdnů | 63% | 54% | 73% |
| Týden 104 | 56% | 49% | 69% |
| ACR50 | |||
| 52 týdnů | 46% | 41% | 62% |
| Týden 104 | 43% | 37% | 59% |
| ACR70 | |||
| 52 týdnů | 27% | 26% | 46% |
| Týden 104 | 28% | 28% | 47% |
| Hlavní klinická odpověď a | 28% | 25% | 49% |
| a Hlavní klinická reakce je definována jako dosažení odpovědi ACR70 za nepřetržité šestiměsíční období b p <0.05 Humira/Mtx vs. MTX for ACR 20 p <0.001 Humira/Mtx vs. MTX for ACR 50 a 70 a Hlavní klinická odpověď c p <0.001 Humira/Mtx vs. Humira |
V týdnu 52 Všechny jednotlivé složky kritérií odezvy ACR pro studii RA-V se zlepšily ve skupině Humira/MTX a zlepšení byla udržována na týden 104.
Radiografická odezva
Ve studii strukturální poškození RA-III bylo radiograficky hodnoceno a vyjádřeno jako změna v celkovém ostrém skóre (TSS) a jeho složky skóre eroze a skóre společného prostoru (JSN) ve 12. měsíci ve srovnání s výchozím hodnotou. Na začátku byla medián TSS přibližně 55 v placebu a 40 mg každý druhý týden. Výsledky jsou uvedeny v tabulce 6. Pacienti ošetřeni HUMIRA/MTX prokázali menší radiografickou progresi než pacienti, kteří dostávali samotné MTX po 52 týdnech.
Tabulka 6: Radiografické průměrné změny během 12 měsíců ve studii RA-III
| Placebo/Mtx | Humira/Mtx 40 mg každý druhý týden | Placebo/Mtx-Humira/Mtx (95% Confidence Interval*) | P-hodnota ** | |
| Celkové ostré skóre | 2.7 | 0.1 | 2.6 (1.4 3,8) | <0.001 |
| Skóre eroze | 1.6 | 0.0 | 1,6 (NULL,9 2.2) | <0.001 |
| Skóre JSN | 1.0 | 0.1 | 0,9 (NULL,3 1,4) | 0.002 |
| *95% intervaly spolehlivosti pro rozdíly ve skóre změn mezi MTX a Humirou. ** Na základě analýzy pořadí |
V otevřeném prodloužení studie RA-III 77% původních pacientů léčených jakoukoli dávkou humiry bylo radiograficky hodnoceno po 2 letech. Pacienti udržovali inhibici strukturálního poškození měřenou pomocí TSS. Padesát čtyři procent nemělo progresi strukturálního poškození, jak je definováno změnou v TSS nulové nebo méně. Padesát pět procent (55%) pacientů původně léčených 40 mg Humirou každý druhý týden bylo radiograficky hodnoceno po 5 letech. Pacienti pokračovali v inhibici strukturálního poškození 50% nevykazovali žádnou progresi strukturálního poškození definovaného změnou v TSS nulové nebo méně.
Ve studii bylo hodnoceno poškození strukturálních kloubů RA-V jako ve studii RA-III. Větší inhibice radiografické progrese, jak je hodnoceno změnami skóre erozí TSS a JSN, byla pozorována v kombinované skupině Humira/MTX ve srovnání s monoterapií MTX nebo Humira v 52. týdnu a také v týdnu 104 (viz tabulka 7).
Tabulka 7: Radiografická průměrná změna* ve studii RA-V
| MTX a N = 257 | Humira ab N = 274 | Humira/Mtx N = 268 | ||
| 52 týdnů | Celkové ostré skóre | 5.7 (4.2 7.3) | 3.0 (1.7 4.3) | 1.3 (NULL,5 2.1) |
| Skóre eroze | 3.7 (2.7 4,8) | 1.7 (1.0 2.4) | 0,8 (NULL,4 1,2) | |
| Skóre JSN | 2.0 (1.2 2.8) | 1.3 (NULL,5 2.1) | 0,5 (NULL,0 1,0) | |
| 104 týdnů | Celkové ostré skóre | 10.4 (7.7 13.2) | 5,5 (3.6 7.4) | 1,9 (NULL,9 2,9) |
| Skóre eroze | 6.4 (4.6 8.2) | 3.0 (2.0 4.0) | 1,0 (NULL,4 1,6) | |
| Skóre JSN | 4.1 (2.7 5.4) | 2.6 (NULL,5 3.7) | 0,9 (NULL,3 1,5) | |
| * Průměr (95% interval spolehlivosti) a p <0.001 Humira/Mtx vs. MTX at 52 a 104 weeks a for Humira/Mtx vs. Humira at 104 weeks b p <0.01 for Humira/Mtx vs. Humira at 52 týdnů |
Reakce fyzické funkce
Ve studiích Ra-I až IV Humira ukázal výrazně větší zlepšení než placebo v indexu zdravotního postižení dotazníku pro hodnocení zdraví (HAQ-DI) od základní linie do konce studie a výrazně větší zlepšení než placebo ve zdravotnických výkonech, jak bylo hodnoceno v průzkumu zdraví krátké formy (SF 36). Zlepšení bylo pozorováno jak v souhrnu fyzické složky (PCS), tak v souhrnu mentálních složek (MCS).
Ve studii RA-III bylo průměrné (95% CI) zlepšení v HAQ-di od základní linie v týdnu 52 0,60 (NULL,55 0,65) u pacientů s humirou a 0,25 (NULL,17 0,33) pro placebo/mtx (P. <0.001) patients. Sixty-three percent of Humira-treated patients achieved a 0.5 or greater improvement in HAQ-DI at week 52 in the double-blind portion of the study. Eighty-two percent of these patients maintained that improvement through week 104 a a similar proportion of patients maintained this response through week 260 (5 years) of open-label treatment. Mean improvement in the SF-36 was maintained through the end of measurement at week 156 (3 years).
Ve studii Ra-V vykázaly Haq-di a fyzická složka SF-36 větší zlepšení (P <0.001) for the Humira/Mtx combination therapy group versus either the MTX moneherapy or the Humira moneherapy group at 52 týdnů which was maintained through Týden 104.
Juvenilní idiopatická artritida
Bezpečnost a účinnost Humira byla hodnocena ve dvou studiích (studie JIA-I a JIA-II) u pacientů s aktivní polyartikulární juvenilní idiopatickou artritidou (JIA).
Studujte jia-i
Bezpečnost a účinnost Humira byla hodnocena v multicentrické randomizované studii s dvojitě zaslepením paralelní skupiny u 171 pacientů, kteří byli 4 až 17 let věku s polyartikulární JIA. Ve studii byli pacienti stratifikováni do dvou skupin: MTX ošetřeno nebo nemtx léčeno. Všichni pacienti museli vykazovat známky aktivního mírného nebo závažného onemocnění navzdory předchozí léčbě analgetiky NSAIDS Kortikosteroidy nebo DMARD. Ze studie byli vyloučeni pacienti, kteří byli předchozí léčbou s jakýmikoli biologickými DMARD.
Studie zahrnovala čtyři fáze: otevřený vedení ve fázi (OL-LI; 16 týdnů) dvojitě slepá randomizovaná fáze stažení (DB; 32 týdnů) otevřenou fázi prodloužení (OLE-BSA; až 136 týdnů) a fáze pevné dávky (ole-Fd; 16 týdnů). V prvních třech fázích studie byla Humira podávána na základě povrchu těla v dávce 24 mg/m² až do maximální celkové dávky těla 40 mg subkutánně (SC) každý druhý týden. Ve fázi ole-FD byli pacienti léčeni 20 mg Humirou SC každý druhý týden, pokud jejich hmotnost byla menší než 30 kg a s 40 mg Humira SC každý druhý týden, pokud jejich hmotnost byla 30 kg nebo větší. Pacienti zůstali na stabilních dávkách NSAID a nebo prednisonu (≤ 0,2 mg/kg/den nebo 10 mg/den maximum).
Pacienti prokazující pediatrickou odpověď ACR 30 na konci fáze OL-LI byli randomizováni do fáze dvojité blind (DB) studie a dostávali Humiru nebo placebo každý druhý týden po dobu 32 týdnů nebo do světla onemocnění. Vzplanutí onemocnění bylo definováno jako zhoršení ≥ 30% z výchozí hodnoty v ≥ 3 ze 6 dětských jádrových kritérií ACR ≥ 2 aktivních kloubů a zlepšení> 30% v nejvýše 1 ze 6 kritérií. Po 32 týdnech nebo v době vzplanutí onemocnění během fáze DB byli léčeni ve fázi prodloužení s otevřenou značkou na základě režimu BSA (OLEBSA) před přeměnou na pevný režim dávky na základě tělesné hmotnosti (ole-FD fáze).
Studujte jia-i Klinická odpověď
Na konci 16-týdenní fáze OL-LI 94% pacientů ve vrstvě MTX a 74% pacientů v non-MTX Stratum byli pediatričtí respondenti ACR 30. Ve fázi DB významně méně pacientů, kteří dostávali humiru, zažili vzplanutí onemocnění ve srovnání s placebem bez MTX (43% vs. 71%) as MTX (37% vs. 65%). Více pacientů léčených Humirou nadále vykazovalo pediatrické odpovědi ACR 30/50/70 v týdnu 48 ve srovnání s pacienty léčenými placebem. Pediatrické odpovědi ACR byly udržovány až dva roky ve fázi OLE u pacientů, kteří dostávali Humiru během studie.
Studujte jia-iI
Humira was assessed in an open-label multicenter study in 32 patients who were 2 to <4 years of age or 4 years of age a older weighing <15 kg with moderately to severely active polyarticular JIA. Most patients (97%) received at least 24 týdnů of Humira treatment dosed 24 mg/m² up to a maximum of 20 mg každý druhý týden as a single SC injection up to a maximum of 120 weeks duration. During the study most patients used concomitant MTX with fewer reporting use of corticosteroids or NSAIDs. The primary objective of the study was evaluation of safety [see Nežádoucí účinky ].
Psoriatická artritida
Bezpečnost a účinnost Humira byla hodnocena ve dvou randomizovaných dvojitě slepých placebech kontrolovaných studiích u 413 pacientů s psoriatickou artritidou (PSA). Po dokončení obou studií 383 pacientů zapsaných do otevřené studie prodloužení, ve které bylo 40 mg Humira podáno každý druhý týden.
Studie PSA-I zařadila 313 dospělých pacientů s mírně až vážně aktivním PSA (> 3 oteklé a> 3 něžné klouby), kteří měli nedostatečnou reakci na terapii NSAID v jedné z následujících forem: (1) distální interfalangální (DIP) zapojení (n = 23); (2) polyartikulární artritida (absence revmatoidních uzlů a přítomnost plakové psoriázy) (n = 210); (3) mutiláni artritidy (n = 1); (4) asymetrický PSA (n = 77); nebo (5) as (n = 2). Pacienti na terapii MTX (158 z 313 pacientů) při zápisu (stabilní dávka ≤ 30 mg/týden po dobu> 1 měsíce) by mohli pokračovat v MTX ve stejné dávce. Během 24týdenního dvojitého období studie byly podávány dávky humira 40 mg nebo placeba každý druhý týden.
Ve srovnání s léčbou placebem Humirou vedlo ke zlepšení měření aktivity onemocnění (viz tabulky 8 a 9). U pacientů s PSA, kteří obdrželi Humiru, byly klinické reakce patrné u některých pacientů v době první návštěvy (dva týdny) a v probíhající otevřené studii byli udržováni až 88 týdnů. Podobné odpovědi byly pozorovány u pacientů s každým z podtypů psoriatické artritidy, ačkoli jen málo pacientů bylo zaregistrováno do artritidy mulilan a ankylozující spondylitidní podtypy. Odpovědi byly podobné u pacientů, kteří byli nebo nedostávali souběžnou terapii MTX na začátku.
Pacienti s psoriatickým postižením nejméně tří procent povrchu těla (BSA) byli hodnoceni na odpovědi psoriatické plochy a indexu závažnosti (PASI). Po 24 týdnech byly proporce pacientů, kteří dosáhli 75% nebo 90% zlepšení v PASI, 59%, respektive 42% ve skupině Humira (n = 69) ve srovnání s 1% a 0% ve skupině s placebem (n = 69) (P. <0.001). PASI responses were apparent in some patients at the time of the first visit (two weeks). Odpověďs were similar in patients who were or were ne receiving concomitant MTX therapy at baseline.
Tabulka 8: Reakce ACR ve studii PSA-I (procento pacientů)
| Placebo N = 162 | Humira* N = 151 | |
| ACR20 | ||
| 12. týden | 14% | 58% |
| 24. týden | 15% | 57% |
| ACR50 | ||
| 12. týden | 4% | 36% |
| 24. týden | 6% | 39% |
| ACR70 | ||
| 12. týden | 1% | 20% |
| 24. týden | 1% | 23% |
| * P. <0.001 for all comparisons between Humira a placebo |
Tabulka 9: Složky aktivity onemocnění ve studii PSA-I
| Parametr: medián | Placebo N = 162 | Humira* N = 151 | ||
| Základní linie | 24 týdnů | Základní linie | 24 týdnů | |
| Počet jemných kloubů a | 23.0 | 17.0 | 20.0 | 5.0 |
| Počet oteklých kloubů b | 11.0 | 9.0 | 11.0 | 3.0 |
| Globální hodnocení lékaře c | 53.0 | 49.0 | 55.0 | 16.0 |
| Globální hodnocení pacienta c | 49.5 | 49.0 | 48.0 | 20.0 |
| Bolest c | 49.0 | 49.0 | 54.0 | 20.0 |
| Index postižení (HAQ) d | 1.0 | 0.9 | 1.0 | 0.4 |
| CRP (MG/DL) e | 0.8 | 0.7 | 0.8 | 0.2 |
| * P. <0.001 for Humira vs. placebo comparisons based on median changes a Měřítko 0-78 b Měřítko 0-76 c Vizuální analogová stupnice; 0 = nejlepší 100 = nejhorší d Index zdravotního postižení dotazníku pro hodnocení zdraví; 0 = nejlepší 3 = nejhorší; Měří schopnost pacienta provádět následující: Dress/Groom Arise Eat Walk Reach Uchopení udržuje hygienu a udržuje každodenní aktivitu. e Normální rozsah: 0-0,287 mg/dl |
Podobné výsledky byly pozorovány u další 12týdenní studie u 100 pacientů se střední až těžkou psoriatickou artritidou, kteří měli suboptimální odpověď na DMARD terapii, jak se projevily ≥ 3 něžnými klouby a ≥ 3 oteklé klouby při zápisu.
Radiografická odezva
Ve studiích PSA byly hodnoceny radiografické změny. Rentgenové snímky zápěstí a nohou rukou byly získány na začátku a 24. týdnu během dvojitého slepého období, kdy byli pacienti na humiru nebo placebu a ve 48 týdnech, kdy byli všichni pacienti na otevřené štítku Humira. Modifikované celkové ostré skóre (MTSS), které zahrnovalo distální interfalangální klouby (tj. Nedoběžné s TSS používanými pro revmatoidní artritidu), bylo použito pro posouzení rentgenových snímků slepých do léčebné skupiny.
Humira-treated patients demonstrated greater inhibition of radiographic progression compačervený to placebo-treated patients a this effect was maintained at 48 týdnů (see Table 10).
Tabulka 10: Změna modifikovaného celkového ostrého skóre u psoriatické artritidy
| Placebo N = 141 | Humira N = 133 | ||
| 24. týden | 24. týden | Týden 48 | |
| Základní linie mean | 22.1 | 23.4 | 23.4 |
| Průměrná změna ± SD | 0,9 ± 3,1 | -0,1 ± 1,7 | -0,2 ± 4,9* |
| * <0.001 for the difference between Humira Týden 48 a Placebo 24. týden (primary analysis) |
Reakce fyzické funkce
Ve studii byly fyzické funkce a postižení PSA-I hodnoceny pomocí indexu HAQ Index Desability (HAQ-DI) a zdravotního průzkumu SF-36. Pacienti léčeni 40 mg Humirou každý druhý týden vykazovali větší zlepšení oproti základní linii ve skóre HAQ-Di (průměrné pokles o 47% a 49% ve 12. a 24. týdnu) ve srovnání s placebem (průměrné pokles o 1% a 3% ve 12. týdnu). V týdnech 12 a 24 pacienti léčených Humirou vykazovali větší zlepšení ze základní linie v souhrnu fyzikální složky SF-36 ve srovnání s pacienty léčenými placebem a žádné zhoršení v souhrnném skóre mentální složky SF-36. Zlepšení fyzické funkce založené na HAQ-DI bylo udržováno až 84 týdnů prostřednictvím otevřené části studie.
Ankylozující spondylitida
Bezpečnost a účinnost HUMIRA 40 mg každý druhý týden byla hodnocena u 315 dospělých pacientů u randomizované 24týdenní dvojité placebem kontrolované studie u pacientů s aktivní ankylozující spondylitidou (AS), kteří měli nedostatečnou odpověď na glukokortikoidy NSAID analgetiky methotrexát. Aktivní, jak bylo definováno jako pacienti, kteří splnili nejméně dvě z následujících tří kritérií: (1) Bath jako index aktivity onemocnění (Basdai) skóre ≥ 4 cm (2) vizuální analogové skóre (VAS) pro celkovou bolest zad ≥ 40 mm a (3) ranní tuhost ≥ 1 hodina. Po oslepeném období následovalo období otevřené značky, během kterého pacienti dostávali Humiru 40 mg každý druhý týden subkutánně po dobu dalších 28 týdnů.
Zlepšení měření aktivity onemocnění bylo poprvé pozorováno ve 2. týdnu a udržováno po dobu 24 týdnů, jak je znázorněno na obrázku 2 a tabulce 11.
Odpověďs of patients with total spinal ankylosis (n=11) were similar to those without total ankylosis.
Obrázek 2: ASAS 20 Reakce od návštěvy studie AS-I
 |
Ve 12 týdnech bylo ASAS 20/50/70 odpovědí dosaženo o 58% 38%, respektive 23% u pacientů, kteří dostávali Humiru, ve srovnání s 21% 10% a 5% u pacientů, kteří dostávali placebo (P <0.001). Similar responses were seen at 24. týden a were sustained in patients receiving open-label Humira for up to 52 týdnů.
Větší část pacientů léčených Humirou (22%) dosáhla nízké úrovně aktivity onemocnění po 24 týdnech (definované jako hodnota <20 [on a scale of 0 to 100 mm] in each of the four ASAS response parameters) compačervený to patients treated with placebo (6%).
Tabulka 11: Složky ankylozující aktivity onemocnění spondylitidy
| Placebo N = 107 | Humira N = 208 | |||
| Základní linie mean | 24. týden mean | Základní linie mean | 24. týden mean | |
| Křídla 20 kritérií Reakce* | ||||
| Globální hodnocení aktivity nemoci pacienta a * | 65 | 60 | 63 | 38 |
| Celková bolest zad* | 67 | 58 | 65 | 37 |
| Zánět b * | 6.7 | 5.6 | 6.7 | 3.6 |
| Basfi c * | 56 | 51 | 52 | 34 |
| Basdai d skóre* | 6.3 | 5.5 | 6.3 | 3.7 |
| Basmi e skóre* | 4.2 | 4.1 | 3.8 | 3.3 |
| Tragus to Wall (cm) | 15.9 | 15.8 | 15.8 | 15.4 |
| Bederní flexe (cm) | 4.1 | 4.0 | 4.2 | 4.4 |
| Rotace děložního čípku (stupně) | 42.2 | 42.1 | 48.4 | 51.6 |
| Bederní boční flexe (cm) | 8.9 | 9.0 | 9.7 | 11.7 |
| Intermalleolární vzdálenost (cm) | 92.9 | 94.0 | 93.5 | 100.8 |
| CRP f * | 2.2 | 2.0 | 1.8 | 0.6 |
| a Procento subjektů s nejméně 20% a 10. zlepšením měřeno na vizuálním Analogová stupnice (VAS) s 0 = None a 100 = závažné b Průměr otázek 5 a 6 Basdai (definované v â? c Ankylozující spondylitida funkční index spondylitidy d Ankylozující lázeň ankylozující spondylitida index aktivity onemocnění e Ankylozující spondylitida metrologie ankylozující lázeň f C-reaktivní protein (MG/DL) * Statisticky významná pro srovnání mezi Humirou a placebem v 24. týdnu |
Druhá randomizovaná multicentrická dvojitě zaslepená placebem kontrolovaná studie 82 pacientů s ankylozující spondylitidou vykazovala podobné výsledky.
Pacienti léčeni Humirou dosáhli zlepšení ze základní linie v dotazníku dotazníku o kvalitě života (ASQOL) (-3,6 vs. -1.1) a v průzkumu fyzické složky (NULL,4 vs. 1,9) (NULL,4 vs. 1,9) (NULL,4 vs. 1,9) ve srovnání s placebem v týdnu 24.
Nemoc dospělého Crohna
Bezpečnost a účinnost více dávek humira byla hodnocena u dospělých pacientů s mírně až vážně aktivním CD CD CD (Crohn's Index aktivity (CDAI) ≥ 220 a ≤ 450) v randomizovaných dvojitě zaslepených placebem kontrolovaných studiích. Byly povoleny souběžné stabilní dávky aminosalicylátů kortikosteroidů a/nebo imunomodulačních látek a 79% pacientů nadále dostávalo alespoň jeden z těchto léků.
Indukce klinické remise (definované jako CDAI <150) was evaluated in two studies. In Study CD-I 299 TNF-blocker naïve patients were raomized to one of four treatment groups: the placebo group received placebo at Weeks 0 a 2 the 160/80 group received 160 mg Humira at Week 0 a 80 mg at Week 2 the 80/40 group received 80 mg at Week 0 a 40 mg at Week 2 a the 40/20 group received 40 mg at Week 0 a 20 mg at Week 2. Clinical results were assessed at 4. týden.
Ve druhé indukční studii studie CD-II 325 pacientů, kteří ztratili odpověď nebo byli intolerantní na předchozí infliximabovou terapii, byli randomizováni, aby dostávali buď 160 mg humira v týdnu 0 a 80 mg v týdnu 2 nebo placeba v týdnech 0 a 2. Klinické výsledky byly hodnoceny ve 4. týdnu.
Udržování klinické remise bylo hodnoceno ve studii CD-III. V této studii 854 pacientů s aktivním onemocněním dostávalo otevřenou značku Humira 80 mg v týdnu 0 a 40 mg ve 2. týdnu. Pacienti byli poté randomizováni ve 4 až 40 mg humiře každý druhý týden 40 mg humira každý týden nebo placebem. Celková doba trvání studie byla 56 týdnů. Pacienti v klinické odpovědi (snížení CDAI ≥ 70) ve 4. týdnu byli stratifikováni a analyzováni odděleně od pacientů, kteří nejsou v klinické odpovědi ve 4. týdnu.
Indukce klinické remise
Větší procento pacientů léčených 160/80 mg Humirou dosáhlo indukci klinické remise versus placebo ve 4. týdnu bez ohledu na to, zda pacienti byli blokátorem TNF na ¯ve (CD-I) nebo ztratili odpověď na infliximab (CD-II) (viz tabulka 12).
Tabulka 12: Indukce klinické remise ve studiích CD-I a CD-II (procento pacientů)
| CD-I | CD-II | |||
| Placebo N = 74 | Humira 160/80 mg N = 76 | Placebo N = 166 | Humira 160/80 mg N = 159 | |
| 4. týden | ||||
| Klinická remise | 12% | 36%* | 7% | 21%* |
| Klinická odpověď | 34% | 58%** | 34% | 52%** |
| Klinická remise is CDAI score <150; clinical response is decrease in CDAI of at least 70 points. * P. <0.001 for Humira vs. placebo pairwise comparison of proportions ** str <0.01 for Humira vs. placebo pairwise comparison of proportions |
Údržba klinické remise
Ve studii CD-III bylo ve 4. týdnu 58% (499/854) pacientů v klinické odpovědi a byly hodnoceny v primární analýze. V týdnech 26 a 56 větší podíl pacientů, kteří byli v klinické odpovědi ve 4. týdnu, dosáhli klinické remise ve skupině Humira 40 mg každý druhý týden údržba ve srovnání s pacienty ve skupině s údržbou placeba (viz tabulka 13). Skupina, která dostávala terapii Humira každý týden, neprokázala výrazně vyšší míru remise ve srovnání se skupinou, která dostávala Humiru každý druhý týden.
Tabulka 13: Údržba klinické remise u CD-III (procento pacientů)
| Placebo N = 170 | 40 mg humira každý druhý týden N = 172 | |
| 26. týden | ||
| Klinická remise | 17% | 40%* |
| Klinická odpověď | 28% | 54%* |
| Týden 56 | ||
| Klinická remise | 12% | 36%* |
| Klinická odpověď | 18% | 43%* |
| Klinická remise is CDAI score <150; clinical response is decrease in CDAI of at least 70 points. *P. <0.001 for Humira vs. placebo pairwise comparisons of proportions |
Z těch, kteří jsou v reakci na 4. týden, kteří dosáhli remise během studijních pacientů ve skupině Humira každý druhý týden, udržovala remisi delší dobu než pacienti ve skupině s údržbou placeba. Mezi pacienty, kteří nebyli v reakci do 12. týdne terapie pokračovali po 12 týdnech, nevedli k výrazně více odpovědi.
Pediatrická Crohnova nemoc
U 192 pediatrických pacientů (6 až 17 let věku) byla provedena randomizovaná dvojitá slepá 52týdenní klinická studie 2 dávkových koncentrací humir (studie PCD-I) s mírně až vážně aktivním Crohnovým onemocněním (definované jako pediatrické crohnové index onemocnění (PCDAI)> 30). Zapsaní pacienti měli za předchozí dva roky nedostatečnou odpověď na kortikosteroidy nebo imunomodulátor (tj. Azathioprin 6-merkaptopurin nebo methotrexát). Pacienti, kteří dříve obdrželi blokátor TNF, se mohli přihlásit, pokud dříve měli ztrátu odpovědi nebo intolerance na tento blokátor TNF.
Pacienti dostávali otevřenou indukční terapii v dávce na základě jejich tělesné hmotnosti (≥ 40 kg a <40 kg). Patients weighing ≥40 kg received 160 mg (at Week 0) a 80 mg (at Week 2). Patients weighing <40 kg received 80 mg (at Week 0) a 40 mg (at Week 2). At 4. týden patients within each body weight category (≥40 kg a <40 kg) were raomized 1:1 to one of two maintenance dose regimens (high dose a low dose). The high dose was 40 mg každý druhý týden for patients weighing ≥40 kg a 20 mg každý druhý týden for patients weighing <40 kg. The low dose was 20 mg každý druhý týden for patients weighing ≥40 kg a 10 mg každý druhý týden for patients weighing <40 kg.
Během studie byly povoleny doprovodné stabilní dávky kortikosteroidů (prednisonová dávka ≤ 40 mg/den nebo ekvivalent) a imunomodulátory (azathioprin 6-merkaptopurin nebo methotrexát).
Ve 12. týdnu pacienti, kteří zažili vzplanutí onemocnění (zvýšení PCDAI ≥ 15 od 4. týdne a absolutní PCDAI> 30), nebo kteří byli neodpovídajícími (tj. Nedostali se snížením PCDAI ≥ 15 z výchozího po sobě jdoucího po sobě jdoucích návštěv od sebe vzdáleného dávky); Pacienti, kteří eskalovali dávku, byli považováni za selhání léčby.
Na začátku 38% pacientů dostávalo kortikosteroidy a 62% pacientů dostávalo imunomodulátor. Čtyřicet čtyři procent (44%) pacientů dříve ztratilo odpověď nebo netolerantní na blokátor TNF. Střední základní skóre PCDAI bylo 40.
Z 192 pacientů celkem 188 pacientů dokončilo 4 týdny indukční období 152 pacientů dokončilo 26 týdnů léčby a 124 pacientů dokončilo 52 týdnů léčby. Padesát jedna procent (51%) (48/95) pacientů ve skupině s nízkou údržbou dávky eskalovalo dávku a 38% (35/93) pacientů v dávce s vysokou udržovací dávkou.
Ve 4. týdnu bylo 28% (52/188) pacientů v klinické remisi (definováno jako pcdai ≤ 10).
Proporce pacientů při klinické remisi (definované jako PCDAI ≤ 10) a klinická odpověď (definovaná jako snížení PCDAI nejméně 15 bodů od základní linie) byly hodnoceny v týdnu 26 a 52.
V obou týdnech 26 a 52 byl podíl pacientů při klinické remisi a klinické odpovědi numericky vyšší ve skupině s vysokou dávkou ve srovnání se skupinou s nízkou dávkou (tabulka 14). Doporučený režim údržby je 20 mg každý druhý týden u pacientů s vážením <40 kg a 40 mg každý druhý týden for patients weighing ≥ 40 kg. Every week dosing is ne the recommended maintenance dosing regimen [see Dávkování a podávání ].
Tabulka 14: Klinická remise a klinická odpověď ve studii PCD-I
| Dávka s nízkou údržbou † (20 nebo 10 mg každý druhý týden) N = 95 | Dávka s vysokou údržbou N = 93 | |
| 26. týden | ||
| Klinická remise ‡ | 28% | 39% |
| Klinická odpověď§ | 48% | 59% |
| 52 týdnů | ||
| Klinická remise ‡ | 23% | 33% |
| Klinická odpověď§ | 28% | 42% |
| † V dávce nízké údržby byla 20 mg každý druhý týden u pacientů vážících ≥ 40 kg a 10 mg každý druhý týden pro osmičování pacientů <40 kg. <40 kg. ‡ Klinická remise definována jako pcdai ≤ 10. §Klinická odezva definována jako snížení PCDAI nejméně 15 bodů od základní linie. |
Ulcerativní kolitida dospělých
Bezpečnost a účinnost humir byla hodnocena u dospělých pacientů s mírně až vážně aktivní ulcerativní kolitidou (mayo skóre 6 až 12 na 12 bodové stupnici s endoskopickou podřízenou 2 až 3 na stupnici 0 až 3) navzdory souběžnému nebo předchozímu léčbě a imunosupresivami, jako jsou kortikosteroidoidoidy azathioprin a azathioprin a azathioprin a azathioprin a apatroidoids asTronizované a-conntrol-conntrol-conntrol-contorel-conntrol-conntrol-control-control-control-con-control-conling ( UC-II). Obě studie zapsaly pacienty s TNF-blokátorem NA ¯ve, ale studium UC-II také umožnilo vstup pacientů, kteří ztratili odpověď nebo byli intolerantní na TNFBlockers. Čtyřicet procent (40%) pacientů zapsaných do studie UC-II dříve použilo další blokátor TNF.
Byly povoleny souběžné stabilní dávky aminosalicylátů a imunosupresiv. Ve studiích pacienti UC-I a II dostávali aminosalicyláty (69%) kortikosteroidy (59%) a/nebo azathioprin nebo 6-MP (37%) na začátku. V obou studiích dostávalo 92% pacientů alespoň jeden z těchto léků.
V obou studiích byla hodnocena indukce klinické remise (definované jako majonérské skóre ≤ 2 bez jednotlivých subscores> 1) v týdnu. Klinická remise v 52. týdnu a trvalá klinická remise (definovaná jako klinická remise v obou týdnech 8 a 52) byla hodnocena ve studii UC-II.
Ve studii byli pacienti na-blokátoru UC-I 390 TNF-blokátora na ¯ve randomizováni do jedné ze tří léčených skupin pro analýzu primární účinnosti. Skupina placeba obdržela placebo v týdnech 0 2 4 a 6. Skupina 160/80 obdržela 160 mg humiru v týdnu 0 a 80 mg v týdnu 2 a 80/40 skupina obdržela 80 mg humira v týdnu 0 a 40 mg v týdnu 2. týden v týdnu 2 v obou humárenských skupinách dostávalo každý další týden.
Ve studii bylo 518 pacientů UC-II randomizováno, aby dostávali buď Humiru 160 mg v týdnu 0 80 mg ve 2. a 40 mg každý druhý týden počínaje týdnem až 50. týdnem nebo placebem začínajícím v týdnu 0 a každý druhý týden až 50. týden. Kortikosteroidní kužel byl povolen od 8. týdne.
V obou studiích UC-I a UC-II je větší procento pacientů léčených 160/80 mg Humirou ve srovnání s pacienty léčenými placebem dosaženým indukcí klinické remise. Ve studii UC-II je větší procento pacientů léčených 160/80 mg Humirou ve srovnání s pacienty léčenými placebem dosaženo trvalé klinické remisi (klinická remise v obou týdnech 8 a 52) (tabulka 15).
Tabulka 15: Indukce klinické remise ve studiích UC-I a UC-II a trvalá klinická remise ve studii UC-II (procento pacientů)
| Studujte uc-i | Studujte uc-iI | |||||
| Placebo N = 130 | Humira 160/80 mg N = 130 | Rozdíl léčby (95% CI) | Placebo N = 246 | Humira 160/80 mg N = 248 | Rozdíl léčby (95% CI) | |
| Indukce klinické remise (klinická remise v 8. týdnu) | 9,2% | 18,5% | 9,3%* (NULL,9%17,6%) | 9,3% | 16,5% | 7,2%* (NULL,2%12,9%) |
| Trvalá klinická remise (klinická remise v obou týdnech 8 a 52) | N/a | N/a | N/a | 4,1% | 8,5% | 4,4%* (NULL,1%8,6%) |
| Klinická remise je definována jako mayo skóre ≤ 2 bez jednotlivých subscores> 1. CI = interval spolehlivosti * P. <0.05 for Humira vs. placebo pairwise comparison of proportions |
Ve studii UC-I nebyl v 8. týdnu pozorován statisticky významný rozdíl v klinické remisi mezi skupinou Humira 80/40 mg a skupinou placeba.
Ve studii bylo UC-II 17,3% (43/248) ve skupině Humira v klinické remisi v týdnu 52 ve srovnání s 8,5% (21/246) ve skupině s placebem (rozdíl léčby: 8,8%; 95% interval spolehlivosti (CI): [2,8% 14,5%]; P P; P; P; P; P; P; P; P; P; P; P; <0.05).
V podskupině pacientů ve studii UC-II s předchozím blokátorem TNF používá rozdíl léčby pro indukci klinické remise je nižší než u celé studijní populace a rozdíly v léčbě pro trvalou klinickou remisi a klinickou remisi v 52 se zdály být podobné těm, které byly pozorovány v celé studijní populaci. Podskupina pacientů s předchozím používáním blokátoru TNF dosáhla indukci klinické remise při 9% (9/98) ve skupině Humira oproti 7% (7/101) ve skupině s placebem a udržovala klinickou remisi při 5% (5/98) ve skupině Humira oproti 1% (1/101) ve skupině placebo. V podskupině pacientů s předchozím blokátorem TNF bylo použití 10% (10/98) v klinické remisi v 52. týdnu ve skupině Humira oproti 3% (3/101) ve skupině s placebem.
Pediatrická ulcerózní kolitida
The safety and efficacy of HUMIRA were assessed in a multicenter randomized double-blind trial (Study PUC-I NCT02065557) in 93 pediatric patients 5 years to 17 years of age with moderately to severely active ulcerative colitis (Mayo score 6 to 12 with endoscopy subscore of 2 to 3 points confirmed by centrally read endoscopy) who had an inadequate response or intolerance to therapy with kortikosteroidy a/nebo imunomodulátor (tj. Azathioprin 6mercaptopurin nebo methotrexát). Patnáct z 93 pacientů (16%) ve studii mělo předchozí zkušenosti s blokátorem TNF. Pacienti, kteří dostávali kortikosteroidy při zápisu, bylo povoleno zužovat jejich kortikosteroidní terapii po 4. týdnu.
Sedmdesát sedm pacientů bylo zpočátku randomizováno 3: 2, aby bylo dosaženo dvojitě slepé léčby s jednou ze dvou dávek humiry. Pacienti v obou dávkovacích skupinách dostávali 2,4 mg/kg (maximálně 160 mg) v týdnu 0 1,2 mg/kg (maximálně 80 mg) v týdnu 2 a 0,6 mg/kg (maximálně 40 mg) ve 4 a 6. týdnech, které byly také připsané a přičítaly se také přičítač a přičítaly a přičítaly a přičítaly a přičítaly a přičítaly a přičítaly a přičítaly a přičítaly a přičítaly a přičítaly se a přijímaly a přičítaly se 160 mg/kg (maximálně 160 mg). Otevřeno s otevřenou značkou s Humirou při vyšší dávce.
At Week 8 62 patients who demonstrated clinical response per Partial Mayo Score (PMS; a subset of the Mayo score with no endoscopic component and defined as a decrease in PMS ≥ 2 points and ≥ 30% from baseline) were randomized equally to receive double-blind treatment with HUMIRA 0.6 mg/kg (maximum of 40 mg) every other week (lower dosage group) or 0.6 mg/kg (maximum of 40 mg) every week (Skupina vyšší dávky). Před změnou návrhu studie bylo 12 pacientů, kteří prokázali klinickou odpověď na PMS, randomizovali se pro přijetí placeba.
Neexistují žádné očekávané klinicky relevantní rozdíly v účinnosti mezi studovaným vyšším dávkováním podávané během 52týdenního pokusu PUC-I a doporučenou dávkou Humiry [viz viz Dávkování a podávání Klinická farmakologie ].
Pacienti, kteří splnili kritéria pro vzplanutí onemocnění ve 12. týdnu nebo po 12. týdnu, byli randomizováni, aby dostali sobovou dávku 2,4 mg/kg (maximálně 160 mg) nebo dávku 0,6 mg/kg (maximálně 40 mg) a poté pokračovali v dávce, do níž byly randomizovány v 8. týdnu.
Koncové koncové body studie byly klinické remise na PMS (definované jako PMS ≤ 2 a žádné individuální subkore> 1) v 8. týdnu a klinická remise podle skóre Mayo (definované jako mayo skóre ≤ 2 a žádné individuální subkore> 1) v 52. týdnu u pacientů, kteří dosáhli klinické reakce na PMS 8. týdne. 30% z výchozí hodnoty) v 52. týdnu v 8. týdnu Respondenti PMS Endoskopické zlepšení (definované jako mayo endoskopické subkore ≤ 1) v týdnu 52 v 8. týdnu respondentů PMS a Mayo skóre remise v týdnu 52 v 8. týdne pms pmitters.
Výsledky 8. týdne
V 8. týdnu bylo remise PMS dosaženo o 60% [28/47; 95% interval spolehlivosti (CI): (44% 74%)] pacientů ve skupině s vyšším dávkováním (bez 16 pacientů dostávajících otevřenou dávkování) a 43% [13/30; 95% CI: (25% 63%)] pacientů ve skupině s nižším dávkováním. Výsledky skupiny s vyšším dávkováním jsou reprezentativní pro očekávané výsledky s doporučenou dávkou [viz Dávkování a podávání Klinická farmakologie ].
52 týdnů Results
V 52. týdnu byly hodnoceny koncové body u populace pacientů, kteří dostávali dvojitě slepé placebo humira 0,6 mg/kg (maximálně 40 mg) každý druhý týden nebo Humira 0,6 mg/kg (maximálně 40 mg) každý týden mezi 8. a týdnem 52 (tabulka 16).
Tabulka 16: Klinická remise klinická odpověď a endoskopické zlepšení v 52. týdnu u pediatrických pacientů s ulcerativní kolitidou (studie PUC-1)
| Placebo a N/N (%) 95% CI | Humira Maximum of 40 mg (0.6 mg/kg) every other week b N/N (%) 95% CI | Humira Maximum of 40 mg (0.6 mg/kg) every week c N/N (%) 95% CI | |
| Klinická remise in Week 8 PMS responders | 4/12 (33%) (10%65%) | 9/31 (29%) (14%48%) | 14/31 (45%) (27%64%) |
| Klinická odpověď in Week 8 PMS responders | 4/12 (33%) (10%65%) | 19/31 (61%) (42%78%) | 21/31 (68%) (49%83%) |
| Endoskopické zlepšení v 8. týdnu Respondenti PMS | 4/12 (33%) (10%65%) | 12/31 (39%) (22%58%) | 16/31 (52%) (33%70%) |
| Klinická remise in Week 8 PMS remitters | 3/8 (38%) (9%76%) | 9/21 (43%) (22%66%) | 10/22 (45%) (24%68%) |
| CI = interval spolehlivosti a Dvanáct pacientů, kteří prokázali klinickou odpověď na PMS v 8. týdnu, bylo randomizováno pro přijetí placeba. Existují omezení pro interpretabilitu dat placeba v důsledku malé velikosti vzorku. b Dávka každý druhý týden studovaná během 52týdenního pokusu PUC-I je nižší dávka než doporučená dávka Humira [viz Dávkování a podávání ]. c Neexistují žádné očekávané klinicky relevantní rozdíly v účinnosti mezi studovaným vyšší dávkou podávanou během 52týdenního pokusu PUC-I a doporučenou dávkou Humiry. Poznámka: Pacienti s chybějícími hodnotami v 52. týdnu nebo kteří byli randomizováni, aby byli podrobeni léčbě redukcí nebo údržbě v důsledku vzplanutí onemocnění, byli považováni za nereagující za koncové body 52 týdnů. |
Plakesová psoriáza
Bezpečnost a účinnost humir byla hodnocena v randomizovaných dvojitě slepých placebokontrolních studiích u 1696 dospělých subjektů se středně těžkou až závažnou chronickou plakovou psoriázou (PS), kteří byli kandidáty na systémovou terapii nebo fototerapii.
Studie PS-I vyhodnotila 1212 subjektů s chronickým PS s ≥ 10% povrchovou plochou těla (BSA) zapojení lékařského globálního hodnocení (PGA) alespoň mírné závažnosti onemocnění a plochou psoriázy a index závažnosti (PASI) ≥12. V období subjekty obdržely placebo nebo humiru v počáteční dávce 80 mg v týdnu 0, po které následovala dávka 40 mg každý druhý týden počínaje 1. týdnem. Po 16 týdnech léčby subjektů, kteří dosáhli alespoň PASI 75 odpovědi v 16. týdnu definovaném jako PASI skóre zlepšení nejméně 75% relativní k výchozímu zadání B a podroben B a otevřeným týdnemi B a otevřený týden B a podrobeno B a Open BOGEL HUMIRA.
Po 17 týdnech subjektů s otevřenou terapií štítků, kteří udržovali alespoň PASI 75 odpověď v 33. týdnu a byli původně randomizováni na aktivní terapii v období A, byly znovu randomizovány v období C, aby přijaly 40 mg humira každý druhý týden nebo placebo po dobu dalších 19 týdnů. Ve všech léčebných skupinách bylo průměrné základní skóre PASI 19 a globální skóre globálního hodnocení lékaře se pohybovalo od mírného (53%) až po závažné (41%) až po velmi závažné (6%).
Studie PS-II vyhodnotila 99 subjektů randomizovaných na Humiru a 48 subjektů randomizovaných na placebo s chronickou plakovou psoriázou s ≥ 10% BSA a PASI ≥12. Subjekty obdržely placebo nebo počáteční dávku 80 mg humira v týdnu 0, po které následuje 40 mg každý druhý týden počínaje 1. týdnem po dobu 16 týdnů. Ve všech léčebných skupinách bylo průměrné základní skóre PASI 21 a skóre základního PGA se pohybovalo od mírného (41%) až po závažné (51%) až po velmi závažné (8%).
Studie PS-I a II vyhodnotily podíl subjektů, které dosáhly jasného nebo minimálního onemocnění na 6-bodové stupnici PGA, a podíl subjektů, které dosáhly snížení skóre PASI nejméně 75% (PASI 75) z výchozího hodnoty v 16. týdnu (viz tabulka 17 a 18).
Studie navíc hodnotila podíl subjektů, které udržovaly PGA jasné nebo minimální nemoci nebo PASI 75 reakci po 33. týdnu a před 52. týdnem.
Tabulka 17: Výsledky účinnosti po 16 týdnech ve studii PS-I Počet subjektů (%)
| Humira 40 mg každý druhý týden N = 814 | Placebo N = 398 | |
| PGA: Jasné nebo minimální * | 506 (62%) | 17 (4%) |
| Po 75 | 578 (71%) | 26 (7%) |
| * Clear = žádná nadmořská výška plaku Žádná stupnice plus nebo mínus hyperpigmentace nebo difúzní růžová nebo červená zbarvení minimální = je možné, ale je obtížné zjistit, zda existuje mírné zvýšení plaku nad normální kůží plus nebo mínus povrchové suchost s nějakým bílým zbarvením plus až do červeného zbarvení |
Tabulka 18: Výsledky účinnosti po 16 týdnech ve studii PS-II Počet subjektů (%)
| Humira 40 mg každý druhý týden N = 99 | Placebo N = 48 | |
| PGA: Jasné nebo minimální * | 70 (71%) | 5 (10%) |
| Po 75 | 77 (78%) | 9 (19%) |
| * Clear = žádná nadmořská výška plaku Žádná stupnice plus nebo mínus hyperpigmentace nebo difúzní růžová nebo červená zbarvení Minimální = možné, ale obtížně zjistit, zda existuje mírné zvýšení plaku nad normální kůží plus nebo mínus povrchová suchost s nějakým bílým zbarvením plus nebo mínus až do červeného zbarvení |
Navíc ve studii subjekty PS-I na Humiru, kteří udržovali Pasi 75, byli znovu rerandomizováni na humiru (n = 250) nebo placebo (n = 240) v týdnu 33. Po 52 týdnech léčby s humirou více subjektů na humiru ve srovnání s subjekty, kteří byli znovu-randomizováni na placebo na placebo na placebo na základě placebo na placebo na placebo na základě placebo na placebo (68% vs. 28%) (79% vs. 43%).
Celkem 347 stabilních respondentů se zúčastnilo hodnocení stažení a retrénování ve studii s otevřenou značkou. Střední čas na relaps (pokles na PGA mírný nebo horší) byl přibližně 5 měsíců. Během období odstoupení žádný subjekt nezaznamenal transformaci na pustulární nebo erytrodermickou psoriázu. Celkem 178 subjektů, které relapsovaly znovu iniciované léčby s 80 mg Humirou, pak 40 mg každý druhý týden počínaje 1. týdnem. V 16. týdnu 69% (123/178) subjektů mělo reakci na PGA jasnou nebo minimální.
Randomizovaná dvojitá slepá studie (studie PS-III) porovnávala účinnost a bezpečnost humira versus placebo u 217 dospělých subjektů. Subjects in the study had to have chronic plaque psoriasis of at least moderate severity on the PGA scale fingernail involvement of at least moderate severity on a 5-point Physician’s Global Assessment of Fingernail Psoriasis (PGA-F) scale a Modified Nail Psoriasis Severity Index (mNAPSI) score for the target-fingernail of ≥ 8 and either a BSA involvement of at least 10% nebo BSA zapojení nejméně 5% s celkovým skóre MNAPSI pro všechny nehty ≥ 20. Subjekty dostaly počáteční dávku 80 mg humira, po které následovala 40 mg každý druhý týden (počínaje týdnem po počáteční dávce) nebo placebo po dobu 26 týdnů po dobu dalších 26 týdnů. Tato studie vyhodnotila podíl subjektů, které dosáhly jasného nebo minimálního posouzení s alespoň 2 stupňovým zlepšením na stupnici PGA-F a podílem subjektů, které dosáhly alespoň 75% zlepšení od výchozí hodnoty v skóre MNAPSI (MNAPSI 75) v 26. týdnu.
V 26. týdnu dosáhla vyšší podíl subjektů ve skupině Humira než ve skupině s placebem koncovým bodem PGA-F. Dále vyšší podíl subjektů ve skupině Humira než ve skupině s placebem dosáhl MNAPSI 75 v 26. týdnu (viz tabulka 19).
Tabulka 19: Výsledky účinnosti po 26 týdnech
| Koncový bod | Humira 40 mg každý druhý týden* N = 109 | Placebo N = 108 |
| PGA-F: ≥ 2 stupně a jasné nebo minimální | 49% | 7% |
| MNAPS 75 | 47% | 3% |
| *Subjekty obdržely 80 mg Humiry v 0. týdnu následované 40 mg každý druhý týden počínaje 1. týdnem. |
Rovněž byla hodnocena bolest nehtů a ve studii PS-III bylo pozorováno zlepšení bolesti nehtů.
Hidradenitis Supsurative
Dvě randomizované dvojitě zaslepené placebem kontrolované studie (studie HS-I a II) hodnotily bezpečnost a účinnost humir u celkem 633 dospělých subjektů se středně těžkou až závažnou suidradenitidou (HS) s Hurley Stage II nebo III onemocnění V obou studiích subjekty obdržely placebo nebo humiru v počáteční dávce 160 mg v týdnu 0 80 mg ve 2. a 40 mg každý týden počínaje 4. týdnem a pokračovaly do 11. týdne. Subjekty používaly lokální antiseptické promývání denně. Současné použití perorálních antibiotik bylo povoleno ve studii HS-II.
Obě studie hodnotily klinickou odpověď Hidradenitis Supsurativa (HISCR) ve 12. týdnu. HISCR byla definována jako alespoň 50% snížení celkového abscesu a počtu zánětlivých uzlů bez zvýšení počtu abscesů a bez zvýšení počtu vyčerpávající píštěle vzhledem k základní linii (viz tabulka 18). Snížení bolesti kůže související s HS bylo hodnoceno pomocí numerické stupnice hodnocení u pacientů, kteří do studie vstoupili s počátečním výchozím skóre 3 nebo více na 11 bodové stupnici.
V obou studiích dosáhl vyšší podíl subjektů ošetřených placebem než placebem (viz tabulka 20).
Tabulka 20: Výsledky účinnosti po 12 týdnech u subjektů s mírnou až těžkou hidradenitidou Supsurativa
| Studie HS i | Studie HS iI* | |||
| Placebo | Humira 40 mg Weekly | Placebo | Humira 40 mg Weekly | |
| Hidradenitis Supsurative Klinická odpověď (HiSCR) | N = 154 40 (26%) | N = 153 64 (42%) | N = 163 45 (28%) | N = 163 96 (59%) |
| *19,3% subjektů ve studii HS-II pokračovalo v základní perorální antibiotické terapii během studie. |
V obou studiích od 12. do 35. týdne (období B) byly subjekty, které obdržely Humiru, znovu-randomizovány na 1 ze 3 léčebných skupin (Humira 40 mg každý týden Humira 40 mg každý druhý týden nebo placebo). Subjekty, které byly randomizovány do placeba, byly přiděleny k přijímání Humira 40 mg každý týden (studie HS-I) nebo placebo (studie HS-II).
Během období B vzplanutí HS definované jako ≥ 25% se zvýšilo z výchozí hodnoty u abscesů a počtu zánětlivých uzlů a s minimálně 2 dalšími lézemi bylo zdokumentováno u 22 (22%) ze 100 subjektů, které byly staženy z humiry po léčbě HUMIRA po primárním časovém úseku ve dvou studiích.
Dospělý uveitida
Bezpečnost a účinnost humir byla hodnocena u dospělých pacientů s neinfekční meziprodukt a panuveitidou bez pacientů s izolovanou přední uveitidou ve dvou randomizovaných dvojitě maskovaných placebem kontrolovaných studiích (UV I a II). Pacienti dostávali placebo nebo humiru v počáteční dávce 80 mg následované 40 mg každý druhý týden počínaje týden po počáteční dávce. Koncovým bodem primární účinnosti v obou studiích byl čas na selhání léčby.
Selhání léčby bylo vícesložkovým výsledkem definovaným jako vývoj nových zánětlivých chorioretinálních a/nebo zánětlivých vaskulárních lézí sítnice Zvýšení stupně přední komory (AC) buněčného stupně (VH) nebo snížení nejlépe korigované vizuální ostrosti (BCVA).
Studie UV I hodnotila 217 pacientů s aktivní uveitidou při léčbě kortikosteroidy (perorální prednison v dávce 10 až 60 mg/den). Všichni pacienti dostávali standardizovanou dávku prednisonu 60 mg/den při vstupu do studie, po kterém následoval povinný rozvrh zúžení s úplným přerušením kortikosteroidů do 15. týdne.
Studie UV II hodnotila 226 pacientů s neaktivní uveitidou, zatímco byla léčena kortikosteroidy (perorální prednison 10 až 35 mg/den) na začátku, aby byla kontrola jejich onemocnění. Pacienti následně podstoupili povinný rozvrh zúžení s úplným přerušením kortikosteroidů do 19. týdne.
Klinická odpověď
Výsledky obou studií prokázaly statisticky významné snížení rizika selhání léčby u pacientů léčených Humirou versus pacienty, kteří dostávali placebo. V obou studiích všechny složky primárního koncového bodu kumulativně přispívaly k celkovému rozdílu mezi Humirou a placebem (tabulka 21).
Tabulka 21: Čas do selhání léčby ve studiích UV I a UV II
| UV i | UV iI | |||||
| Placebo (N = 107) | Humira (N = 110) | HR [95% CI] a | Placebo (N = 111) | Humira (N = 115) | HR [95% CI] a | |
| Selhání b n (%) | 84 (NULL,5) | 60 (NULL,5) | 0.50 [0,36 0,70] | 61 (NULL,0) | 45 (39.1) | 0.57 [0,39 0,84] |
| Střední čas do selhání (měsíce) [95% CI] | 3.0 [2.7 3.7] | 5.6 [3.9 9.2] | N/a | 8.3 [4,8 12.0] | NE c | N/a |
| a HR Humira versus placebo z proporcionálních rizik regrese s léčbou jako faktorem. b Selhání léčby v 6. týdnu nebo po 6. týdnu ve studii UV I nebo ve 2. týdnu nebo po 2. týdnu ve studii UV II se počítá jako událost. Subjekty, které studii ukončily studii, byly cenzurovány v době vypadnutí. c NE = ne estimable. Fewer than half of at-risk subjects had an event. |
Obrázek 3: Křivky Kaplan-Meier shrnující čas na selhání léčby 6. týdne (studium UV I) nebo 2. týden (studie UV II)
 |
Poznámka: str
Dětská uveitida
Bezpečnost a účinnost Humiry byla hodnocena v randomizované dvojitě maskované placebocověné studii 90 pediatrických pacientů od 2 do 2 do 2 do 2 do 2 do 2 do 2 do 2 do 2 do 2 do <18 years of age with active JIA-associated non-infectious uveitis (PUV-I). Patients received either placebo or 20 mg Adalimumab (if < 30 kg) or 40 mg Adalimumab (if ≥ 30 kg) every other week in combination with a dose of methotrexate. Concomitant dosages of corticosteroids were permitted at study entry followed by a maatory červenýuction in topical corticosteroids within 3 months.
Primárním koncovým bodem byl „čas na selhání léčby“. Kritéria určující selhání léčby byly zhoršení nebo trvalé nezvýšení při očním zánětu nebo zhoršení očních komorbidit.
Klinická odpověď
Humira significantly decreased the risk of treatment failure by 75% relative to placebo (HR = 0.25 [95% CI: 0.12 0.49]) (Table 22).
Tabulka 22: Výsledky analýzy času na selhání léčby (studie PUV-I)
| Placebo (N = 30) | Humira (N = 60) | HR (95% CI) a | |
| Selhání (n[%]) | 18 (60%) | 16 (NULL,7%) | 0,25 (NULL,12 0,49) |
| Střední čas do selhání (týdny) (95% CI) b | 24.1 (NULL,4 81.0) | NEC | |
| a HR adalimumabu versus placebo z proporcionálních rizik regrese s léčbou jako faktorem. b Odhaduje se na základě Kaplan-Meierovy křivky. c NE = ne estimable. Fewer than half of at-risk subjects had an event. |
Obrázek 4: Kaplan-Meier křivky shrnující čas na selhání léčby (studie PUV-I)
 |
Studujte puv-i
Poznámka: p = placebo (číslo rizika); H = humira (rizikové číslo).
Reference
1. Institut pro rakovinu. Program dozoru epidemiologie a konečných výsledků (SEER). Incidence SEER INCIDICE SURDE MASE 17 Registry 2000-2007.
Informace o pacientovi pro Humiru
Humira ®
(Hu-Mare-ah)
(Adalimumab)
injekce pro subkutánní použití
Přečtěte si průvodce léky, který přichází s Humirou, než začnete užívat a pokaždé, když dostanete doplnění. Mohou existovat nové informace. Tento průvodce medikací nezabírá místo rozhovoru se svým lékařem o vašem zdravotním stavu nebo léčbě.
Jaké jsou nejdůležitější informace, které bych měl vědět o Humiře?
Humira is a medicine that affects your immune system. Humira can lower the ability of your immune system to fight infections. U lidí, kteří užívají Humiru, došlo k vážným infekcím. Tyto vážné infekce zahrnují tuberkulózu (TB) a infekce způsobené viry houbami nebo bakteriemi, které se šířily po celém těle. Někteří lidé zemřeli na tyto infekce.
- Před zahájením HUMIRA by vás měl váš lékař vyzkoušet na TB.
- Váš lékař by vás měl pečlivě zkontrolovat, zda nedošlo k příznakům a příznakům TB během léčby Humirou.
Neměli byste začít užívat Humiru, pokud máte nějakou infekci, pokud váš lékař neříká, že je to v pořádku.
Před zahájením HUMIRA sdělte svému lékaři, pokud:
- Myslíte si, že máte infekci nebo máte příznaky infekce, jako například:
- horečka potí nebo zimnice
- Bolesti svalů
- kašel
- dušnost
- Krev v Plegmu
- teplá červená nebo bolestivá kůže nebo vředy na vašem těle
- průjem nebo bolest žaludku
- pálení, když močíte nebo močíte častěji než obvykle
- cítit se velmi unavený
- hubnutí
- jsou léčeny pro infekci.
- Získejte spoustu infekcí nebo mají infekce, které se stále vracejí.
- mít cukrovku.
- mají TB nebo byli v úzkém kontaktu s někým s TB.
- narodili se v židle nebo cestovali do zemí, kde je větší riziko získání TB. Zeptejte se svého lékaře, pokud si nejste jisti.
- Žijte nebo žili v určitých částech země (jako jsou údolí Ohio a údolí řeky Mississippi), kde existuje zvýšené riziko pro získání určitých druhů plísňových infekcí (histoplazmóza kokcidioidomykózy nebo blastomykózy). K těmto infekcím se může stát nebo se stane závažnějšími, pokud používáte Humiru. Zeptejte se svého lékaře, zda nevíte, zda jste žili v oblasti, kde jsou tyto infekce běžné.
- mít nebo mít hepatitidu B.
- Použijte léky Orencia (Abatacept) Kineret (rituximab) nebo purinethol (6-merkaptopurin 6-MP).
- jsou naplánovány na velkou operaci.
Po zahájení HUMIRA okamžitě zavolejte svého lékaře Pokud máte infekci nebo jakékoli známky infekce.
Humira can make you more likely to get infections or make any infection that you may have worse.
Rakovina
- U dětí a dospělých, kteří užívají faktor nekrózy nádoru (TNF)-blokátory, včetně Humira, se může zvýšit šance na získání rakoviny.
- U dětí teenagerů a mladých dospělých používají případy neobvyklých rakovin pomocí blokátorů TNF.
- Lidé s revmatoidní artritidou (RA) Obzvláště vážnější RA mohou mít vyšší šanci na získání jakéhokoli rakoviny zvaného lymfom.
- Pokud používáte blokátory TNF, včetně Humira, vaše šance na získání dvou typů rakoviny kůže se může zvýšit (rakovina bazálních buněk a spinocelulární rakovina kůže). Tyto typy rakoviny obecně neohrožují život, pokud jsou léčeny. Řekněte svému lékaři, pokud máte ránu nebo otevřenou bolest, která se nezhojí.
- Někteří lidé, kteří dostávají blokátory TNF, včetně Humira, vyvinul vzácný typ rakoviny zvané hepatosplenický lymfom T-buněk. Tento typ rakoviny často vede k smrti. Většina z těchto lidí byli teenageři nebo mladí muži. Také většina lidí byla léčena Crohnovou chorobou nebo ulcerózní kolitidou s jiným lékem zvaným imuran (azathioprin) nebo purinetol (6-merkaptopurin 6-MP).
Co je to Humira?
Humira is a medicine called a Tumor Necrosis Factor (TNF) blocker. Humira is used:
- Snížit příznaky a příznaky:
- Mírné až těžké RA u dospělých. Humira can be used alone with methotrexate or with certain other medicines.
- Mírná až těžká polyartikulární mladistvá idiopatická artritida (JIA) u dětí 2 roky a starší. Humira lze použít samostatně nebo s methotrexátem.
- Psoriatická artritida (PSA) u dospělých. Humira can be used alone or with certain other medicines.
- Ankylozující spondylitida (AS) u dospělých.
- Mírná až těžká hidradenitida Supsurativa (HS) u lidí 12 let.
- Léčit střední až těžkou Crohnovu chorobu (CD) u dospělých a dětí ve věku 6 let a starších.
- Léčit střední až těžkou ulcerózní kolitidu (UC) u dospělých a dětí ve věku 5 let a starších. Není známo, zda je Humira účinná u lidí, kteří přestali reagovat nebo nemohli tolerovat léky blokátorů TNF.
- Léčit mírnou až těžkou chronickou (trvající dlouhou dobu) psoriázu plaku (PS) u dospělých kteří mají stav v mnoha oblastech jejich těla a kteří mohou těžit z přijímání injekcí nebo pilulek (systémová terapie) nebo fototerapie (léčba pomocí ultrafialového světla samotného nebo pilulky).
- Léčit neinfekční střední a panuveitidu u dospělých a dětí ve věku 2 let a starší.
Co bych měl říct svému lékaři, než si vezmu Humiru?
Humira may ne be right for you. Before starting Humira tell your doctor about all of your medical conditions including if you:
- mít infekci. Vidět Jaké jsou nejdůležitější informace, které bych měl vědět o Humiře?
- mít nebo mít rakovinu.
- mít jakoukoli necitlivost nebo brnění nebo mít onemocnění, které ovlivňuje váš nervový systém, jako je roztroušená skleróza nebo Guillain-Barré syndrom.
- mít nebo mít srdeční selhání.
- nedávno obdrželi nebo jsou naplánovány na obdržení vakcíny. Při používání Humira můžete dostávat vakcíny s výjimkou živých vakcín. Děti by měly být před zahájením HUMIRA upřenitelné děti se všemi vakcínami.
- jsou alergické na gumu nebo latex. Řekněte svému lékaři, pokud máte nějaké alergie na gumu nebo latex.
- Kryt jehly pro HUMIRA pero 40 mg/0,8 ml Humira 40 mg/0,8 ml předplněné stříkačky HUMIRA 20 mg/0,4 ml předplněné stříkačky a humira 10 mg/0,2 ml předběžnou stříkačku může obsahovat přirozenou kaučuk nebo latex.
- The black needle cover for the HUMIRA Pen 80 mg/0.8 mL HUMIRA 80 mg/0.8 mL prefilled syringe HUMIRA Pen 40 mg/0.4 mL HUMIRA 40 mg/0.4 mL prefilled syringe HUMIRA 20 mg/0.2 mL prefilled syringe HUMIRA 10 mg/0.1 mL prefilled syringe and the vial stopper on the HUMIRA institutional use vial are not Vyrobeno z přírodní gumy nebo latexu.
- jsou alergičtí na humiru nebo na některou z jejích složek. Seznam ingrediencí v Humiru naleznete na konci této medikační příručky.
- jsou těhotné nebo plánují otěhotnět kojení nebo plánují kojit. Vy a váš lékař byste se měli rozhodnout, zda byste měli vzít Humiru, když jste těhotná nebo kojení.
- Mějte dítě a během těhotenství jste používali Humiru. Řekněte lékaře svého dítěte, než vaše dítě obdrží jakékoli vakcíny.
Řekněte svému lékaři o všech lécích, které užíváte včetně předpisu a over-the-theCounter Medicines Vitamins a bylinné doplňky.
Zejména řekněte svému lékaři, pokud používáte:
- Orencia (abatacept) Kineret (Anakinra) Remicade (infliximab) enbrel (etanercept) Cimzia (Certolizumab pegol) nebo Simponi (Golimumab), protože byste neměli používat Humira, když také používáte jeden z těchto léků.
- Rituxan (rituximab). Váš lékař vám nemusí chtít dát Humiru, pokud jste nedávno obdrželi Rituxan (rituximab).
- Imuran (azathioprin) nebo purinethol (6-merkaptopurin 6-MP).
Udržujte s sebou seznam svých léků, abyste mohli ukázat svému lékaři a lékárníkovi pokaždé, když získáte nový lék.
Jak mám vzít Humiru?
- Humira is given by an injection under the skin. Your doctor will tell you how often to take an injection of Humira. This is based on your condition to be treated. Nepřidávejte Humiru častěji, než jste byli předepsáni.
- Úplné pokyny pro přípravu a injekci Humira naleznete v pokynech pro použití uvnitř kartonu.
- Než to uděláte sami, ujistěte se, že vám bylo ukázáno, jak vložit Humiru. Pokud máte nějaké dotazy ohledně injekce, můžete zavolat svému lékaři nebo 1-800-4HUMIRA (1-800-448-6472). Někdo, koho znáte, vám může také pomoci s vaší injekcí poté, co jim bylo ukázáno, jak připravit a vstříknout Humiru.
- Ne Pokuste se vstříknout Humiru, dokud vám nebyla ukázána správný způsob, jak dát injekce. Pokud se váš lékař rozhodne, že vy nebo pečovatel budete moci doma poskytnout své injekce Humiry, měli byste absolvovat školení správným způsobem k přípravě a injekci Humira.
- Ne miss any doses of Humira unless your doctor says it is okay. If you forget to take Humira inject a dose as soon as you remember. Then take your next dose at your regular scheduled time. This will put you back on schedule. In case you are ne sure when to inject Humira call your doctor or pharmacist.
- Pokud si vezmete více Humiru, než vám bylo řečeno, abyste zavolali svého lékaře.
Jaké jsou možné vedlejší účinky Humiry?
Humira can cause serious side effects including:
Vidíte, jaké jsou nejdůležitější informace, které bych měl vědět o Humiru?
- Vážné infekce.
Váš lékař vás prozkoumá pro TB a proveďte test, abyste zjistili, zda máte TB. Pokud se váš lékař domnívá, že jste ohroženi TBC, můžete být léčeni medicínou pro TBC před zahájením léčby Humirou a během léčby Humirou. I když je váš test TB negativní, váš lékař by vás měl pečlivě sledovat, pokud jde o infekce TB, zatímco berete Humiru. Lidé, kteří měli negativní test na kůži TB před přijetím Humira, se vyvinuli aktivní TB. Řekněte svému lékaři, pokud máte některý z následujících příznaků při užívání nebo po přijetí HUMIRA:
- kašel that does ne go away
- Horečka nízkého stupně
- hubnutí
- Ztráta tělesného tuku a svalu (plýtvání)
- Infekce hepatitidy B u lidí, kteří nesou virus v krvi.
Pokud jste nosičem viru hepatitidy B (virus, který ovlivňuje játra), může být virus aktivní, když používáte Humiru. Váš lékař by měl provádět krevní testy před zahájením léčby, když používáte Humiru a několik měsíců po zastavení léčby Humirou. Řekněte svému lékaři, pokud máte některý z následujících příznaků možné infekce hepatitidy B:
- Bolesti svalů
- cítit se velmi unavený
- Tmavá moč
- Kůže nebo oči vypadají žlutě
- malá nebo žádná chuť
- zvracení
- hliněné pohyby střev
- horečka
- zimnice
- nepohodlí žaludku
- Skin vyrážka
- Alergické reakce. U lidí, kteří používají Humiru, se mohou vyskytnout alergické reakce. Pokud máte některý z těchto příznaků vážné alergické reakce, zavolejte svému lékaři nebo si okamžitě získejte lékařskou pomoc:
- kopřivka
- potíže s dýcháním
- Otok očí obličeje rty nebo ústa
- Problémy nervového systému. Mezi příznaky a příznaky problému nervového systému patří: otupělost nebo brnění se slabostí vaší vidění v pažích nebo nohou a závratě.
- Problémy s krví. Vaše tělo nemusí vyrábět dostatek krevních buněk, které pomáhají bojovat proti infekcím nebo pomáhají zastavit krvácení. Mezi příznaky patří horečka, která neodchází modřina nebo krvácení velmi snadno nebo vypadají velmi bledě.
- Nové srdeční selhání nebo zhoršení srdečního selhání, které již máte. Okamžitě zavolejte svého lékaře Pokud získáte nové zhoršující se příznaky srdečního selhání při užívání humiry včetně:
- dušnost
- náhlý přírůstek hmotnosti
- Otok vašich kotníků nebo nohou
- Imunitní reakce včetně syndromu podobného lupusu. Mezi příznaky patří nepohodlí na hrudi nebo bolest, která nezmizí dušnost bolesti dechu nebo vyrážka na tvářích nebo pažích, která se na slunci zhoršuje. Když zastavíte Humiru, mohou se příznaky zlepšit.
- Problémy s jatery. U lidí, kteří používají léky na blokování TNF. Tyto problémy mohou vést k selhání jater a smrti. Pokud máte některý z těchto příznaků, zavolejte svému lékaři ihned:
- cítit se velmi unavený
- špatná chuť k jídlu nebo zvracení
- Kůže nebo oči vypadají žlutě
- Bolest na pravé straně žaludku (břicho)
- Psoriáza. Někteří lidé používající Humiru měli novou psoriázu nebo zhoršení psoriázy, kterou již měli. Řekněte svému lékaři, pokud vyvinete červené šupinaté skvrny nebo zvednuté hrboly, které jsou naplněny hnisem. Váš lékař se může rozhodnout zastavit léčbu Humirou.
Pokud vyvinete některý z výše uvedených příznaků, zavolejte svému lékaři nebo se okamžitě dostanete. Vaše léčba Humirou může být zastavena.
Mezi nejčastější vedlejší účinky Humira patří:
- Reakce místa vstřikování: svědění nebo modřiny zarudnutí vyrážky. Tyto příznaky obvykle zmizí během několika dnů. Okamžitě zavolejte svého lékaře, pokud máte zarudnutí bolesti nebo otok kolem místa injekce, které během několika dnů nezmizí nebo se zhoršuje.
- infekce horních cest dýchacích (včetně infekcí sinus).
- bolest hlavys.
- vyrážka.
Nejedná se o všechny možné vedlejší účinky s Humirou. Řekněte svému lékaři, pokud máte jakýkoli vedlejší účinek, který vás vadí nebo že nezmizí. Požádejte o více informací svého lékaře nebo lékárníka.
Zavolejte svého lékaře, kde najdete lékařskou radu ohledně vedlejších účinků. Můžete nahlásit vedlejší účinky FDA na 1800- FDA-1088.
Jak mám ukládat Humiru?
- Humira ukládejte do lednice při 36 ° F až 46 ° F (2 ° C až 8 ° C). Uložte Humiru v původním kartonu, dokud není použita k jeho ochraně před světlem.
- Ne Zmrazení Humira. Ne Použijte Humiru, pokud je zamrzlá, i když byla rozmrazena.
- Chlazená humira může být použita až do data vypršení vypršení vytištěného na podnosovém peru Humira Carton nebo předplněné stříkačky. Nepoužívejte Humiru po datu vypršení platnosti.
- V případě potřeby například při cestování můžete také ukládat Humiru při teplotě místnosti až do 77 ° F (25 ° C) po dobu až 14 dnů. Uložte Humiru v původním kartonu, dokud není použita k jeho ochraně před světlem.
- Vyhoďte Humiru, pokud byla udržována při teplotě místnosti a nebyla použita do 14 dnů.
- Zaznamenejte datum, kdy nejprve odstraníte Humiru z chladničky v prostorech uvedených na kartonu a dávce.
- Neukládejte humiru v extrémním teplu nebo chladu.
- Ne use a Pen or prefilled syringe if the liquid is cloudy discoločervený or has flakes or particles in it.
- Ne drop or crush Humira. The prefilled syringe is glass.
Udržujte injekční zásoby HUMIRA a všechny ostatní léky mimo dosah dětí.
Obecné informace o bezpečném a efektivním používání Humiry.
Léky jsou někdy předepisovány pro jiné účely než ty, které jsou uvedeny v průvodci s léky. Nepoužívejte Humiru pro podmínku, pro kterou nebyla předepsána. Nedávejte Humiru jiným lidem, i když mají stejný stav. Může jim to poškodit. Tento průvodce medikací shrnuje nejdůležitější informace o Humiru. Pokud byste chtěli více informací, promluvte si se svým lékařem. Lékárníka nebo lékaře můžete požádat o informace o Humiru, která je psána pro zdravotnické pracovníky.
Jaké jsou ingredience v Humiru?
Aktivní složka: Adalimumab
Humira Pen 40 mg/0.8 mL Humira 40 mg/0.8 mL prefilled syringe Humira 20 mg/0.4 mL prefilled syringe Humira 10 mg/0.2 mL prefilled syringe a Humira 40 mg/0.8 mL institutional use vial:
Neaktivní ingredience: Kyselina citronová monohydrát dibasic sodný fosfát dihydrát mannitol monobasic sodný fosfát dihydrát polysorbát 80 sodný sodný citrát a voda pro injekci. Pro úpravu pH se přidává hydroxid sodný.
Humira Pen 80 mg/0.8 mL Humira 80 mg/0.8 mL prefilled syringe Humira Pen 40 mg/0,4 ml Humira 40 mg/0,4 ml prefilled syringe Humira 20 mg/0.2 mL prefilled syringe a Humira 10 mg/0.1 mL prefilled syringe:
Neaktivní ingredience: Mannitol Polysorbate 80 a voda pro injekci.
Pokyny pro použití
Humira ®
(Hu-Mare-ah)
(Adalimumab)
40 mg/0,8 ml
Jednodávkové pero
Ne Pokuste se vstříknit Humiru, dokud vám nebyla ukázána správný způsob, jak poskytnout injekce a přečíst si a porozumět těmto pokynům pro použití. Pokud se váš lékař rozhodne, že vy nebo pečovatel budete moci doma poskytnout své injekce Humiry, měli byste absolvovat školení správným způsobem k přípravě a injekci Humira. Je důležité, abyste si přečetli porozumění a postupovali podle těchto pokynů, abyste vložili Humiru správnou cestou. Je také důležité mluvit se svým lékařem, abyste se ujistili, že rozumíte svým pokynům pro dávkování Humira. Chcete -li si zapamatovat, kdy vložit Humiru, můžete svůj kalendář označit předem. Pokud máte vy nebo váš pečovatel jakékoli dotazy ohledně správného způsobu, jak vložit Humiru, zavolejte svému poskytovateli zdravotní péče.
Důležité:
- Ne Použijte Humiru, pokud je zamrzlá, i když byla rozmrazena.
- Humira pero obsahuje sklo. Neklesněte ani nedržte pero, protože sklo uvnitř se může zlomit.
- Každé pero Humira má na sobě 2 čepice. Neodstraňujte šedou víčko (uzávěr
- Když je tlačítko Plum-zbarvené na pero Humira stisknuto, aby se vaše dávka Humira poskytla hlasitý zvuk kliknutí.
- Musíte cvičit injekci HUMIRA se svým lékařem nebo zdravotní sestrou, abyste nebyli tímto kliknutím vyděšeni, když si začnete dávat injekce doma.
- Zvuk hlasitého kliknutí znamená začátek injekce.
- Budete vědět, že injekce skončila, když se žlutý indikátor objeví plně v pohledu okna a přestane se pohybovat.
Viz část níže volaná Příprava humira pero.
Shromážděte zásoby pro injekci
- Pro každou injekci Humira budete potřebovat následující zásoby.
Najděte čistý rovný povrch, na který je zásoby umístěn.
- 1 alkohol
- 1 bavlněná koule nebo podložka gázy (není zahrnuta do kartonu Humira)
- 1 HUMIRA PEN (viz Obrázek a )
- Kontejner pro propíchnutí odolný proti propíchnutí pro likvidaci pero Humira (není zahrnut do vaší humorské kartonu). Viz Jak bych měl zahodit (zlikvidovat) použité humira? část na konci těchto pokynů pro použití.
Pokud pohodlněji vytáhněte Humira pero z chladničky 15 až 30 minut Před injekcí, aby se kapalina dosáhla teploty místnosti. Ne Odstraňte šedou víčko (čepice Ne teplá humira jiným způsobem (například ne zahřejte ji v mikrovlnné troubě nebo v horké vodě).
Pokud nemáte veškeré zásoby, musíte si dát injekci, přejděte do lékárny nebo zavolejte svému lékárníkovi. Obrázek níže ukazuje, jak vypadá Humira pero. Vidět Obrázek a .
Obrázek a
 |
Zkontrolujte zásobník dávky a Humira pero.
Viz Jak mám ukládat Humiru? část na konci těchto pokynů pro použití.
Obrázek b
 |
Obrázek c
 |
Vyberte místo injekce
Připravte místo injekce
Příprava humira pera
Obrázek e
 |
Umístěte pero a injektujte HUMIRA
Obrázek J.
 |
- Ujistěte se, že název HUMIRA se objeví na značce dávky kartonu a na štítku HUMIRA PEN.
- Ne use a Zavolejte Váš lékař nebo lékárník, pokud:
- Pádete nebo rozdrtíte své humirské pero.
- Těsnění na horní nebo dolní části kartonu jsou rozbité nebo chybějící.
- Datum vypršení platnosti na dávkové zásobníku a pero prošlo.
- Humira pero bylo zmrazeno nebo ponecháno za přímého slunečního světla.
- Humira has been kept at room temperature for longer than 14 Dny nebo Humira byly skladovány nad 77 ° F (25 ° C).
- Držte pero s šedou čepicí (uzávěr
- Ujistěte se, že množství kapaliny v peru je na linii výplně nebo v blízkosti linky plnění, které je vidět oknem. Toto je plná dávka Humira, kterou vstřikujete. Vidět Obrázek b .
- Pokud pero nemá celé množství kapaliny ne use that Pen. Zavolejte svému lékárníkovi.
- Otočte pero a držte pero s šedou čepicí (uzávěr Obrázek c .
- Zkontrolujte roztok okny na boku pera, abyste se ujistili, že kapalina je čistá a bezbarvá. Ne use Vaše HUMIRA PEN, pokud je kapalina zakalená zbarvená nebo pokud má v sobě vločky nebo částice. Zavolejte svému lékárníkovi. Je normální vidět jednu nebo více bublin v okně.
- Dobře si umyjte a osušte ruce.
- Vyberte místo injekce na:
Obrázek d
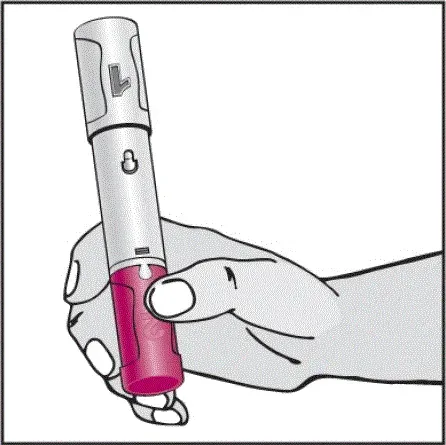
- přední část vašich stehen nebo
- Vaše dolní břicho (břicho). Pokud si vyberete břicho, nepoužívejte plochu 2 palce kolem tlačítka břicha (pupek). Vidět Obrázek d .
- Vyberte jinou stránku pokaždé, když si dáte injekci. Každá nová injekce by měla být podávána alespoň jeden palec od místa, které jste použili předtím.
- Ne vstříkněte Humiru do kůže, která je:
- bolest (nabídka)
- pohmožděné
- červený
- tvrdý
- zjizvené nebo kde máte strie
- Pokud máte psoriázu ne Injekci přímo do jakýchkoli zvednutých silné červené nebo šupinaté kožní skvrn nebo lézí na vaší kůži.
- Ne inject through your clothes.
- Otřete místo injekce přípravou na alkohol (tampon) pomocí kruhového pohybu.
- Ne Před provedením injekce se znovu dotkněte této oblasti. Před injekcí nechte pokožku uschnout. Ne ventilátor nebo foukání na čistou oblast.
- Ne remove the gray cap (Cap # 1) or the plum-coločervený cap (Cap # 2) until right before your injection.
- Držte střed pera (šedé tělo) jednou rukou, abyste se nedotýkali šedé čepice (uzávěr ( Obrázek e .
- Druhou rukou vytáhněte šedou čepici (čepice Obrázek f .
- Vyhoďte šedou čepici (čepice
Obrázek f
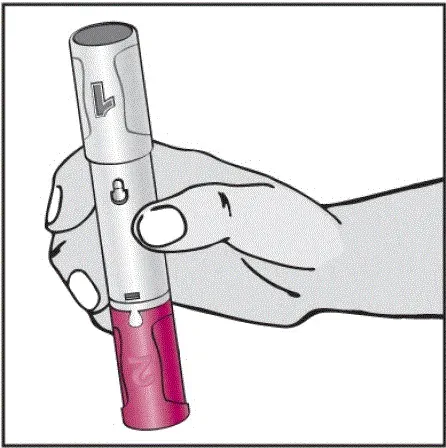
- Ne Dejte šedou čepici (čepice
- Nyní lze vidět rukáv bílé jehly, který zakrývá jehlu.
- Ne Dotkněte se jehly prsty nebo nechte jehlu dotknout se čehokoli.
- Můžete vidět několik kapek kapaliny vycházející z jehly. To je normální.
- Odstraňte švestkově zbarvenou čepici (čepice
Tlačítko aktivátoru plum-zbarveného:
Obrázek g

Obrázek h
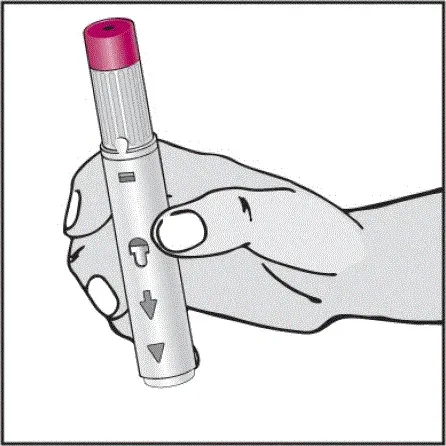
- Ne put the plum-coločervený cap (Cap # 2) back on pero because it could cause medicine to come out of the syringe.
- Otočte pero tak, aby se tlačítko aktivátoru švestkově zbarvilo. Vidět Obrázek g .
- Ne Stisknutím tlačítka aktivátoru je stisknutím, dokud nebudete připraveni vložit Humiru. Stisknutím tlačítka aktivátoru zbarveného švestky uvolní lék z pera.
- Držte pero, abyste mohli vidět okno. Vidět Obrázek h . Je normální vidět jednu nebo více bublin v okně.
- Umístěte pero:
Obrázek i
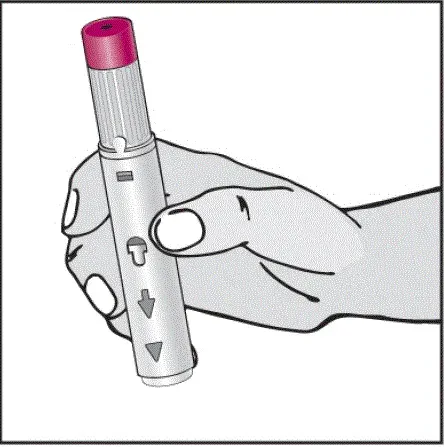
- Sevření Oblast vyčištěné kůže a pevně ji drží, dokud není injekce dokončena. Vidět Obrázek i . Do této zvednuté oblasti kůže se vstříknete.
- Umístěte bílý konec pera rovně (pod úhlem 90 °) a flat against the raised area of your skin that you are squeezing. Místo pero so that it will ne inject the needle into your fingers that are holding the raised skin. Vidět Obrázek J. .
- Injekční HUMIRA
Obrázek k
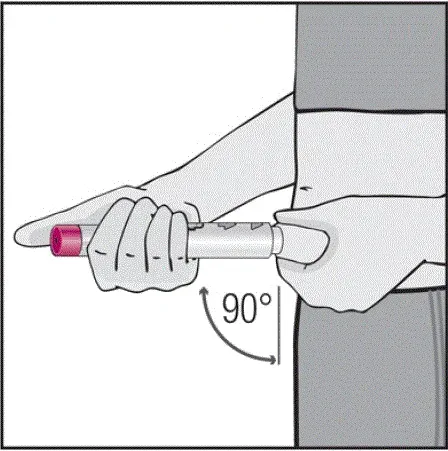
Obrázek l
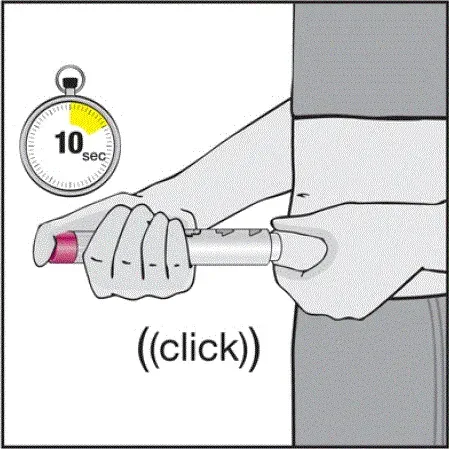
- Je důležité, abyste pero pevně zatlačili před zahájením injekce až proti místa vstřikování.
- Pokračujte dolů zabránit tomu, aby se pero během injekci pohybovalo z kůže.
- Stisknutím tlačítka aktivátoru stisknutím palcem stiskněte injekci. Snažte se nepokrývat okno. Vidět Obrázek k .
- Když stisknete tlačítko aktivátoru Plum-Colored Activator, uslyšíte hlasité „kliknutí“. Hlasité kliknutí znamená začátek injekce.
- Pokračujte v stisknutí tlačítka aktivátoru s švestkem a pokračujte v tlačení pera proti vymaččené zvednuté pokožce, dokud se veškerý lék vstříkne. To může trvat až 10 sekund, takže se pomalu počítá až deset. Po celou dobu neustále tlačíte pero proti vymaččené zvednuté kůži vašeho injekčního místa, abyste získali plnou dávku medicíny.
- Budete vědět, že injekce skončila, když se v pohledu okna plně objeví žlutý indikátor a přestane se pohybovat. Vidět Obrázek l .
- Když je injekce dokončena, pomalu vytáhněte pero z pokožky. Bílý rukáv jehly se bude pohybovat, aby pokryl špičku jehly. Vidět Obrázek m .
Obrázek m
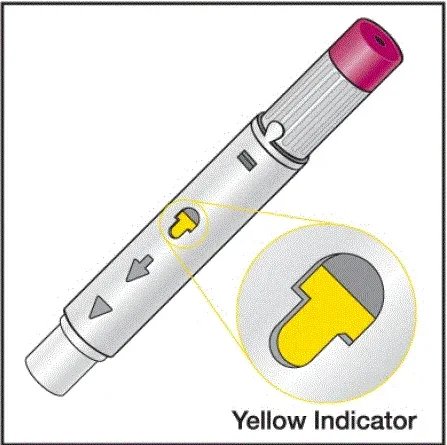
- Ne touch the needle. The white needle sleeve is there to prevent you from touching the needle.
- Na místě injekce může být malé množství kapaliny. To je normální.
- Stiskněte bavlněnou kuličku nebo gázovou podložku na místo vstřikování a držte ji po dobu 10 sekund. Dělat ne Třete místo injekce. Možná máte mírné krvácení. To je normální.
- Vyhoďte (zlikvidujte) své použité humirské pero v nádobě na likvidaci Sharps hned po použití. Viz část Jak bych měl zlikvidovat použité HUMIRA PEN?
- Uchovávejte záznam dat a umístění vašich injekčních webů. Chcete -li si zapamatovat, kdy si vzít Humiru, můžete svůj kalendář označit předem.








Jak bych měl zahodit (zlikvidovat) použité humira?
Obrázek n
 |
- Okamžitě po použití vložte pero do nádoby na likvidaci Sharps pro likvidaci FDA. Vidět Obrázek n . Ne throw away pero in your household tvyrážka.
- Ne try to touch the needle. The white needle sleeve is there to prevent you from touching the needle.
- Pokud nemáte nádobu na likvidaci Sharps Clored FDA, můžete použít kontejner pro domácnost, která je:
- vyrobeno z těžkoprávného plastu
- Lze uzavřít s pevně přiléhajícím víkem odolným proti vpichu, aniž by ostřely mohly vyjít
- Během používání vzpřímeně a stabilní
- odolný vůči únikům a
- Správně označené tak, aby varovala před nebezpečným odpadem uvnitř nádoby.
- Pokud je vaše kontejner pro likvidaci SHARPS téměř plný, budete muset dodržovat své pokyny pro komunitu pro správný způsob, jak zlikvidovat svůj kontejner likvidace Sharps. Mohou existovat státní nebo místní zákony o tom, jak byste měli zahodit použité jehly a stříkačky. Další informace o bezpečném likvidaci Sharps a pro konkrétní informace o likvidaci Sharps ve státě, že žijete na webových stránkách FDA na adrese: https://www.fda.gov/safesharpsdisposal.
- Pro bezpečnost a zdraví vás a ostatních nikdy znovu nepoužívejte HUMIRA pera.
- Použité zásobníky a balení s bavlněnými kuličkami z alkoholu mohou být umístěny do odpadků pro domácnost.
- Ne dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Ne recycle your used sharps disposal container.
- Vždy udržujte nádobu Sharps mimo dosah dětí.
Jak mám ukládat Humiru?
- Humira ukládejte do ledničky mezi 36 ° F až 46 ° F (2 ° C až 8 ° C). Uložte Humiru v původním kartonu, dokud není použita k jeho ochraně před světlem.
- Ne Zmrazení Humira. Ne Použijte Humiru, pokud je zamrzlá, i když byla rozmrazena.
- Chlazená humira může být použita až do data vypršení platnosti vytištěného na podnosu nebo peru Humira Carton. Ne Použijte Humiru po datu vypršení platnosti.
- V případě potřeby například při cestování můžete také ukládat Humiru při teplotě místnosti až do 77 ° F (25 ° C) až pro až do 14 dny. Uložte Humiru v původním kartonu, dokud není použita k jeho ochraně před světlem.
- Odhoďte Humiru, pokud byla udržována při pokojové teplotě a nebyla používána uvnitř 14 dny.
- Zaznamenejte datum, kdy nejprve odstraníte Humiru z chladničky v prostorech uvedených na kartonu a dávce.
- Neukládejte humiru v extrémním teplu nebo chladu.
- Ne use a Pen if the liquid is cloudy discoločervený or has flakes or particles in it.
- Ne drop or crush Humira.
- Udržujte injekční zásoby HUMIRA a všechny ostatní léky mimo dosah dětí.
Pokyny pro použití
Humira ®
(Hu-Mare-ah)
(Adalimumab)
40 mg/0,4 ml
Jednodávkové pero
Před injekcí: Váš poskytovatel zdravotní péče by vám měl ukázat, jak používat Humiru, než ji poprvé použijete. Zavolejte svému poskytovateli zdravotní péče nebo 1-800-4HUMIRA (1-800-448-6472) Pokud potřebujete pomoc.
Obrázek a: Humira Jednodávkové pero
Triamcinolon acetonid Dental Paste USP 0,1
 |
Důležité informace, které potřebujete vědět před injekcí Humira
Ne Použijte pero a zavolejte svého poskytovatele zdravotní péče nebo lékárníka, pokud:
- Kapalina je zakalena zbarvená nebo má vločky nebo částice
- Uběhlo datum vypršení platnosti
- Kapalina byla zamrzlá (i když se rozmrazovala) nebo ponechána na přímém slunečním světle
- Pero bylo upuštěno nebo rozdrceno
Udržujte čepice až těsně před injekcí.
Jak mám ukládat Humiru?
- Humira ukládejte do ledničky mezi 36 ° F až 46 ° F (2 ° C až 8 ° C).
- Uložte Humiru v původním kartonu, dokud není použita k jeho ochraně před světlem.
- Ne freeze
- Chlazená humira může být použita až do data vypršení platnosti vytištěného na podnosu nebo peru Humira Carton.
- V případě potřeby například při cestování můžete také ukládat Humiru při teplotě místnosti až do 77 ° F (25 ° C) až pro až do 14 dny.
- Vyhoďte Humiru, pokud byla udržována při pokojové teplotě a nepoužila se uvnitř 14 dny.
- Zaznamenejte datum, kdy nejprve odstraníte Humiru z chladničky v prostorech uvedených na kartonu a dávce.
- Neukládejte humiru v extrémním teplu nebo chladu.
Udržujte injekční zásoby HUMIRA a všechny ostatní léky mimo dosah dětí.
Před použitím Humira Pen si přečtěte pokyny na všech stránkách
Vzít Humira out of the refrigerator.
Dovolená Humira at room temperature for 15 až 30 minut před injekcí.
- Ne Odstraňte šedou víčko (čepice
- Ne teplé humira jiným způsobem. Například ne zahřejte ji v mikrovlnné troubě nebo v horké vodě.
- Ne Použijte pero, pokud byla tekutina zmrazena (i když se rozmrazí)
 |
Kontrola Datum vypršení platnosti na štítku pera. Ne Pokud uplynulo datum vypršení, použijte pero.
Místo následující na čistém rovném povrchu:
- 1 jednodávkový pero a alkohol
- 1 bavlněná koule nebo podložka gázy (není součástí)
- Kontejner likvidace odolných proti propíchnutí (není součástí). Podívejte se na krok 9 na konci těchto pokynů pro použití pokynů, jak zahodit (zlikvidovat) vaše Humira Pen
Umyjte a osušte vaše ruce.
 |
Vybrat místo injekce:
- Na přední straně stehen nebo
- Vaše břicho (břicho) nejméně 2 palce od pupku (břišní tlačítko)
- Odlišné od vašeho posledního webu injekce
Otřete Místo injekce v kruhovém pohybu s tamponem alkoholu.
- Ne vstříkněte oblečením
- Ne Injekční do kůže, která je bolestivá pohmožděná červená tvrdá zjizvení má strie nebo oblasti s plaky psoriázy
 |
Držet pero s šedou čepicí Kontrola okno.
- Je normální vidět 1 nebo více bublin v okně
- Ujistěte se, že kapalina je čistá a bezbarvá
- Ne Použijte pero, pokud je kapalina zamračená nebo má vločky nebo částice
- Ne Pokud bylo upuštěno nebo rozdrceno, použijte pero
 |
SEM šedá čepice
Vyhoďte čepici pryč.
- Je normální vidět několik kapek tekutiny vycházet z jehly
SEM švestka zbarvená čepice
Vyhoďte čepici pryč.
Otočte pero tak, aby bílá šipka směřovala směrem k injekčnímu místu.
 |
Sevření Kůže v místě vstřikování, aby se vytvořila zvednutá plocha a pevně ji držela, dokud není injekce dokončena.
Bod bílá šipka směrem k injekčnímu místu.
Místo bílý jehlou rukáv rovný (Úhel 90 °) proti injekčnímu místu.
Držet pero tak, abyste viděli inspekční okno.
Ne Stiskněte tlačítko Plum Activator, dokud nebudete připraveni vstoupit.
 |
Je důležité, abyste pero pevně zatlačili před zahájením injekce až proti místa vstřikování.
Pokračujte dolů zabránit tomu, aby se pero během injekci pohybovalo z kůže.
Stiskněte tlačítko Aktivátor švestky a pomalu počítáte 10 sekundy.
- Hlasité kliknutí bude signalizovat start injekce
- Pokračujte v tlaku pero pevně dolů proti místa injekce, dokud není injekce dokončena
- Injekce je dokončena, když se žlutý indikátor přestal pohybovat
 |
Když je injekce dokončena, pomalu vytáhněte pero z kůže. Bílý pouzdro na jehlu zakryje špičku jehly.
- Malé množství kapaliny na místě injekce je normální
Pokud je na volání místa vstřikování více než několik kapek kapaliny 1-800-4HUMIRA (1-800448- 6472) pro pomoc.
Po dokončení vstřikování umístěte bavlněnou kuličku nebo gázovou podložku na kůži místa vstřikování.
- Ne třít
- Mírné krvácení v místě injekce je normální
 |
Jak bych měl zlikvidovat použité HUMIRA PEN?
- Okamžitě po použití vložte použitá jehla a ostřely do FDA vyčištěné nádoby na likvidaci Sharps. Ne throw away (dispose of) loose needles syringes a pero in the household tvyrážka.
- Pokud nemáte kontejner pro likvidaci Sharps FDA, můžete použít kontejner pro domácnost, která je:
- vyrobeno z těžkoprávného plastu
- Lze uzavřít s pevně přiléhajícím víkem odolným proti vpichu, aniž by ostřely mohly vyjít
- Během používání vzpřímeně a stabilní
- odolný vůči únikům a
- Správně označené tak, aby varovala před nebezpečným odpadem uvnitř nádoby.
- Pokud je vaše kontejner pro likvidaci SHARPS téměř plný, budete muset dodržovat své pokyny pro komunitu pro správný způsob, jak zlikvidovat svůj kontejner likvidace Sharps. Mohou existovat státní nebo místní zákony o tom, jak byste měli zahodit použité jehly a stříkačky. Další informace o bezpečné likvidaci Sharps a konkrétních informacích o likvidaci Sharps ve státě, ve kterém žijete na webových stránkách FDA na adrese: https://www.fda.gov/safesharpsdisposal.
- Ne dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Ne recycle your used sharps disposal container.
 |
V domácnosti mohou být umístěny podnos a balení pera upevňuje alkohol tampontu s výtěrem nebo podnosu na gázovou podložku.
Otázky týkající se používání pera Humira
Co když jsem neobdržel osobně školení od poskytovatele zdravotní péče?
- Zavolejte svému poskytovateli zdravotní péče nebo 1-800-4HUMIRA (1-800-448-6472) nebo navštivte www.humira.com, pokud potřebujete pomoc
Jak zjistím, kdy je injekce kompletní?
- Žlutý indikátor se přestal pohybovat. To zabírá 10 sekundy.
Co mám dělat, pokud na místě injekce existuje více než několik kapek tekutiny?
- Volání 1-800-4HUMIRA (1-800-448-6472) pro pomoc
Co když nemám FDA vyčištěný nádoba na likvidaci Sharps nebo správný kontejner pro domácnost?
- Volání 1-800-4HUMIRA (1-800-448-6472) pro bezplatný kontejner s ostře na FDA
Vždy Udržujte pero a nádobu na likvidaci Sharps mimo dosah dětí.
Uchovávejte záznam dat a umístění vašich injekcí. Chcete -li si pamatovat, kdy si vzít HUMIRA označit svůj kalendář předem.
 |
 |
Pokyny pro použití
Humira ®
(Hu-Mare-ah)
(Adalimumab)
Balíčky obsahující 80 mg/0,8 ml
Jednodávkové pero
Před injekcí: Váš poskytovatel zdravotní péče by vám měl ukázat, jak používat Humiru, než ji poprvé použijete. Zavolejte svému poskytovateli zdravotní péče nebo 1-800-4HUMIRA (1-800-448-6472) Pokud potřebujete pomoc.
Obrázek a: Humira Jednodávkové pero
 |
Důležité informace, které potřebujete vědět před injekcí Humira
Ne Použijte pero a zavolejte svého poskytovatele zdravotní péče nebo lékárníka, pokud:
- Kapalina je zakalena zbarvená nebo má vločky nebo částice
- Uběhlo datum vypršení platnosti
- Kapalina byla zamrzlá (i když se rozmrazovala) nebo ponechána na přímém slunečním světle
- Pero bylo upuštěno nebo rozdrceno
Udržujte čepice až těsně před injekcí.
Jak mám ukládat Humiru?
- Humira ukládejte do ledničky mezi 36 ° F až 46 ° F (2 ° C až 8 ° C).
- Uložte Humiru v původním kartonu, dokud není použita k jeho ochraně před světlem.
- Ne freeze
- Chlazená humira může být použita až do data vypršení platnosti vytištěného na podnosu nebo peru Humira Carton.
- V případě potřeby například při cestování můžete také ukládat Humiru při teplotě místnosti až do 77 ° F (25 ° C) až pro až do 14 dny.
- Vyhoďte Humiru, pokud byla udržována při pokojové teplotě a nepoužila se uvnitř 14 dny.
- Zaznamenejte datum, kdy nejprve odstraníte Humiru z chladničky v prostorech uvedených na kartonu a dávce.
- Neukládejte humiru v extrémním teplu nebo chladu.
Udržujte injekční zásoby HUMIRA a všechny ostatní léky mimo dosah dětí.
Před použitím Humira Pen si přečtěte pokyny na všech stránkách
Vzít Humira out of the refrigerator.
Dovolená Humira at room temperature for 15 až 30 minut před injekcí.
- Ne Odstraňte šedou víčko (čepice
- Ne teplé humira jiným způsobem. Například ne zahřejte ji v mikrovlnné troubě nebo v horké vodě
- Ne Použijte pero, pokud byla tekutina zmrazena (i když se rozmrazí)
 |
Kontrola Datum vypršení platnosti na štítku pera. Ne Pokud uplynulo datum vypršení, použijte pero.
Místo následující na čistém rovném povrchu:
- 1 jednodávkový pero a alkohol
- 1 bavlněná koule nebo podložka gázy (není součástí)
- Kontejner likvidace odolných proti propíchnutí (není součástí). Podívejte se na krok 9 na konci těchto pokynů pro použití pokynů, jak zahodit (zlikvidovat) vaše Humira Pen
Umyjte a osušte vaše ruce.
 |
Vybrat místo injekce:
- Na přední straně stehen nebo
- Vaše břicho (břicho) nejméně 2 palce od pupku (břišní tlačítko)
- Odlišné od vašeho posledního webu injekce
Otřete Místo injekce v kruhovém pohybu s tamponem alkoholu.
- Ne vstříkněte oblečením
- Ne Injekční do kůže, která je bolestivá pohmožděná červená tvrdá zjizvení má strie nebo oblasti s plaky psoriázy
 |
Držet pero s šedou čepicí Kontrola okno.
- Je normální vidět 1 nebo více bublin v okně
- Ujistěte se, že kapalina je čistá a bezbarvá
- Ne Použijte pero, pokud je kapalina zamračená nebo má vločky nebo částice
- Ne Pokud bylo upuštěno nebo rozdrceno, použijte pero
 |
SEM šedá čepice
Vyhoďte čepici pryč.
- Je normální vidět několik kapek tekutiny vycházet z jehly
SEM švestka zbarvená čepice
Vyhoďte čepici pryč.
Otočit pero so that the white arrow points toward the injection site.
 |
Sevření Kůže v místě vstřikování, aby se vytvořila zvednutá plocha a pevně ji držela, dokud není injekce dokončena.
Bod bílá šipka směrem k injekčnímu místu.
Místo bílý jehlou rukáv rovný (Úhel 90 °) proti injekčnímu místu.
Držet pero tak, abyste viděli inspekční okno.
 |
Ne Stiskněte tlačítko Plum Activator, dokud nebudete připraveni vstoupit.
Je důležité, abyste pero pevně zatlačili před zahájením injekce až proti místa vstřikování.
Stiskněte tlačítko Aktivátor švestky a pomalu počítáte 15 sekundy.
- Hlasité kliknutí bude signalizovat start injekce
- Pokračujte v tlaku pero pevně dolů proti místa injekce, dokud není injekce dokončena
- Injekce je dokončena, když se žlutý indikátor přestal pohybovat
 |
Když je injekce dokončena, pomalu vytáhněte pero z kůže. Bílý pouzdro na jehlu zakryje špičku jehly.
- Malé množství kapaliny na místě injekce je normální
Pokud je na volání místa vstřikování více než několik kapek kapaliny 1-800-4HUMIRA (1-800448- 6472) pro pomoc.
Po dokončení vstřikování umístěte bavlněnou kuličku nebo gázovou podložku na kůži místa vstřikování.
- Ne třít
- Mírné krvácení v místě injekce je normální
 |
Jak bych měl zlikvidovat použité HUMIRA PEN?
- Okamžitě po použití vložte použitá jehla a ostřely do FDA vyčištěné nádoby na likvidaci Sharps. Ne throw away (dispose of) loose needles syringes a pero in the household tvyrážka.
- Pokud nemáte nádobu na likvidaci Sharps Clored FDA, můžete použít kontejner pro domácnost, která je:
- vyrobeno z těžkoprávného plastu
- Lze uzavřít s pevně přiléhajícím víkem odolným proti vpichu, aniž by ostřely mohly vyjít
- Během používání vzpřímeně a stabilní
- odolný vůči únikům a
- Správně označené tak, aby varovala před nebezpečným odpadem uvnitř nádoby.
- Pokud je vaše kontejner pro likvidaci SHARPS téměř plný, budete muset dodržovat své pokyny pro komunitu pro správný způsob, jak zlikvidovat svůj kontejner likvidace Sharps. Mohou existovat státní nebo místní zákony o tom, jak byste měli zahodit použité jehly a stříkačky. Další informace o bezpečné likvidaci Sharps a konkrétních informacích o likvidaci Sharps ve státě, ve kterém žijete na webových stránkách FDA na adrese: https://www.fda.gov/safesharpsdisposal.
- Ne dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Ne recycle your used sharps disposal container.
 |
V domácnosti mohou být umístěny podnos a balení pera upevňuje alkohol tampontu s výtěrem nebo podnosu na gázovou podložku.
Otázky týkající se používání pera Humira
Co když jsem neobdržel osobní školení od poskytovatele zdravotní péče?
- Zavolejte svému poskytovateli zdravotní péče nebo 1-800-4HUMIRA (1-800-448-6472) nebo navštivte www.humira.com, pokud potřebujete pomoc
Jak zjistím, kdy je injekce kompletní?
- Žlutý indikátor se přestal pohybovat. To zabírá 15 sekundy.
Co mám dělat, pokud na místě injekce existuje více než několik kapek tekutiny?
- Volání 1-800-4HUMIRA (1-800-448-6472) pro pomoc
Co když nemám FDA vyčištěný nádoba na likvidaci Sharps nebo správný kontejner pro domácnost?
- Volání 1-800-4HUMIRA (1-800-448-6472) pro bezplatný kontejner s ostře na FDA
Vždy Udržujte pero a nádobu na likvidaci Sharps mimo dosah dětí.
Uchovávejte záznam dat a umístění vašich injekcí. Chcete -li si pamatovat, kdy si vzít HUMIRA označit svůj kalendář předem.
 |
 |
Pokyny pro použití
Humira ®
(Hu-Mare-ah)
(Adalimumab)
40 mg/0,8 ml 20 MG/0.4 ML AND 10 MG/0.2 ML
Jednorázová předběžná stříkačka
Ne Pokuste se vstříknit Humiru, dokud vám nebyla ukázána správný způsob, jak poskytnout injekce a přečíst si a porozumět těmto pokynům pro použití. Pokud se váš lékař rozhodne, že vy nebo pečovatel budete moci doma poskytnout své injekce Humiry, měli byste absolvovat školení správným způsobem k přípravě a injekci Humira. Je důležité, abyste si přečetli porozumění a postupovali podle těchto pokynů, abyste vložili Humiru správnou cestou. Je také důležité mluvit se svým lékařem, abyste se ujistili, že rozumíte svým pokynům pro dávkování Humira. Chcete -li si zapamatovat, kdy vložit Humiru, můžete svůj kalendář označit předem. Pokud máte vy nebo váš pečovatel jakékoli dotazy ohledně správného způsobu, jak vložit Humiru, zavolejte svému poskytovateli zdravotní péče.
Shromážděte zásoby pro injekci
- Pro každou injekci Humira budete potřebovat následující zásoby.
Najděte čistý rovný povrch, na který je zásoby umístěn.
- 1 alkohol
- 1 bavlněná koule nebo podložka gázy (není zahrnuta do kartonu Humira)
- 1 HUMIRA Preffikovaná stříkačka (viz Obrázek a )
- Kontejner pro propíchnutí odolný proti propíchnutí pro likvidaci injekční stříkačky Humira (není zahrnuta do vaší kartonu Humira). Viz Jak bych měl zahodit (zlikvidovat) použité předplněné stříkačky a jehly? část na konci těchto pokynů pro použití.
Pokud pohodlnější vyjměte svou Humiru předplněnou stříkačku z ledničky 15 až 30 minut Před injekcí, aby se kapalina dosáhla teploty místnosti. Ne Odstraňte kryt jehly a zároveň jíte dosáhnout teploty místnosti. Ne teplá humira jiným způsobem (například ne zahřejte ji v mikrovlnné troubě nebo v horké vodě).
Pokud nemáte veškeré zásoby, musíte si dát injekci, přejděte do lékárny nebo zavolejte svému lékárníkovi.
Obrázek níže ukazuje, jak vypadá předplněná stříkačka. Vidět Obrázek a .
Obrázek a
 |
Kontrola the carton dose tray a prefilled syringe
Viz Jak mám ukládat Humiru? část na konci těchto pokynů pro použití.
Vyberte místo injekce
Připravte místo injekce
Ne ventilátor nebo foukání na čistou oblast.
Připravte stříkačku a jehlu
Obrázek d
 |
Umístěte předběžnou stříkačku a injekci Humiru
Umístěte stříkačku
Obrázek h
 |
Injekční HUMIRA
- Ujistěte se, že název HUMIRA se objeví na dávkovém zásobníku a předběžné etiketě stříkačky.
- Ne use a Zavolejte Váš lékař nebo lékárník, pokud:
- Těsnění na horní nebo dolní části kartonu jsou rozbité nebo chybějící.
- Označování HUMIRA má datum vypršení. Zkontrolujte datum vypršení vypršení na svém kartonu Humira a ne Použijte, pokud datum uplynulo.
- Předplněná stříkačka byla zmrazena nebo ponechána za přímého slunečního světla.
- Humira has been kept at room temperature for longer than 14 Dny nebo Humira byly skladovány nad 77 ° F (25 ° C).
- Kapalina v předplněné stříkačce je zakalená nebo má vločky nebo částice. Ujistěte se, že kapalina je čistá a bezbarvá.
- Dobře si umyjte a osušte ruce.
- Vyberte místo injekce na:
Obrázek b
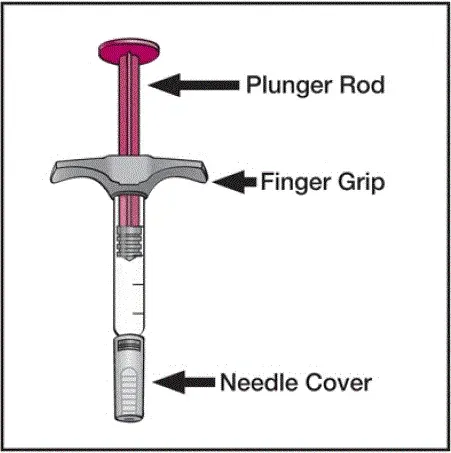
- přední část vašich stehen nebo
- Vaše dolní břicho (břicho). Pokud si vyberete břicho, nepoužívejte plochu 2 palce kolem tlačítka břicha (pupek). Vidět Obrázek b .
- Vyberte jinou stránku pokaždé, když si dáte injekci. Každá nová injekce by měla být podávána alespoň jeden palec od místa, které jste použili předtím.
- Ne vstříkněte do kůže, která je:
- bolest (nabídka)
- pohmožděné
- červený
- tvrdý
- zjizvené nebo kde máte strie
- Pokud máte psoriázu ne Injekci přímo do jakýchkoli zvednutých silné červené nebo šupinaté kožní skvrn nebo lézí na vaší kůži.
- Ne inject through your clothes.
- Otřete místo injekce přípravou na alkohol (tampon) pomocí kruhového pohybu.
- Dělat ne Před provedením injekce se znovu dotkněte této oblasti. Před injekcí nechte pokožku uschnout.
- Kontrola the fluid level in the syringe:
Obrázek c
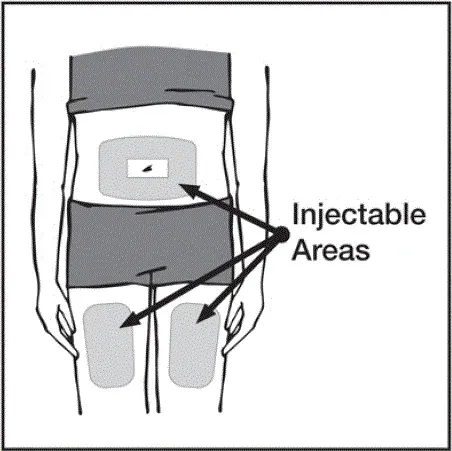
- Držet the syringe with the covečervený needle pointing down. Vidět Obrázek c .
- Držet the syringe at eye level. Look closely to make sure that the amount of liquid in the syringe is the same or close to the:
- 0,8 ml linie pro 40 mg předplněnou stříkačku. Vidět Obrázek d .
- 0,4 ml čáry pro 20 mg předplněnou stříkačku. Vidět Obrázek d .
- 0,2 ml linie pro 10 mg předplněnou stříkačku. Vidět Obrázek d .
- Horní část kapaliny může být zakřivená. Pokud stříkačka nemá správné množství kapaliny ne use that syringe. Zavolejte svému lékárníkovi.
- Odstraňte kryt jehly:
Obrázek e
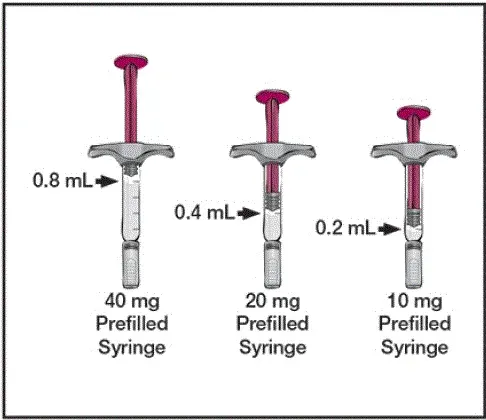
- Držet the syringe in one ha. With the other ha gently remove the needle cover. Vidět Obrázek e .
- Vyhoďte kryt jehly.
- Ne Dotkněte se jehly prsty nebo nechte jehlu dotknout se čehokoli.
- Otočit the syringe so the needle is facing up a hold the syringe at eye level with one ha so you can see the air in the syringe. Pomocí druhé ruky pomalu tlačit Pluntár v tlaku vzduchu jehlou. Vidět Obrázek f .
Obrázek f
- Na konci jehly můžete vidět kapku kapaliny. To je normální.
- Držet the body of the prefilled syringe in one ha between the thumb a index finger. Držet the syringe in your ha like a pencil. Vidět Obrázek g .
Obrázek g
- Ne kdykoli se zatáhněte na píst.
- Druhou rukou jemně stiskněte oblast vyčištěné kůže a pevně ji drží. Vidět Obrázek h .
- Použijte rychlou šipku podobnou pohybu vložte jehlu do stlačené kůže na přibližně a Úhel 45DEGREE. Vidět Obrázek i .
Obrázek i

- Poté, co je jehla v pust kůži. Jemně se zatáhněte na píst.
Pokud se v injekční stříkačce objeví krev:
Obrázek J.
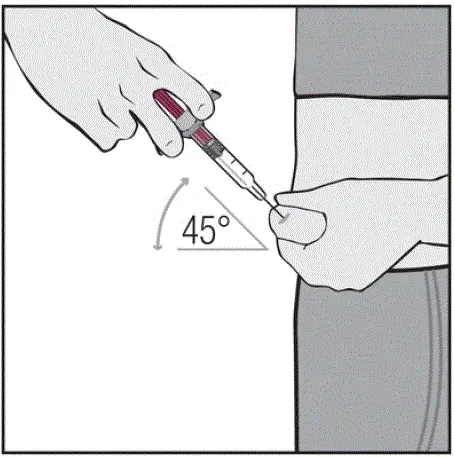
- To znamená, že jste vstoupili do krevní cévy.
- Ne inject Humira.
- SEM the needle out of the skin while keeping the syringe at the same angle.
- Stiskněte a cotton ball or gauze pad over the injection site a hold it for 10 sekundy. Vidět Obrázek J. .
- Ne Opět použijte stejnou stříkačku a jehlu. Zahoďte jehlu a injekční stříkačku do nádoby ostře.
- Ne Třete místo injekce. Možná máte mírné krvácení. To je normální.
- Opakujte kroky 1 až 12 s novou předplněnou stříkačkou.
Pokud se v injekční stříkačce neobjeví žádná krev:
- Pomalu zatlačte píst celou cestu dovnitř, dokud se vstříkne veškerá kapalina a stříkačka je prázdná.
- SEM the needle out of the skin while keeping the syringe at the same angle.
- Stiskněte bavlněnou kuličku nebo gázovou podložku na místo vstřikování a držte ji po dobu 10 sekund. Dělat ne Třete místo injekce. Možná máte mírné krvácení. To je normální.







- Okamžitě po použití zahoďte použité předplněné stříkačky a jehlu v nádobě na likvidaci Sharps. Vidět Jak bych měl zahodit (zlikvidovat) používané předplněné stříkačky a jehly?
- Uchovávejte záznam dat a umístění vašich injekčních webů. Chcete -li si zapamatovat, kdy si vzít Humiru, můžete svůj kalendář označit předem.
Jak bych měl zahodit (zlikvidovat) používané předplněné stříkačky a jehly?
Obrázek k
 |
- Po použití vložte použité jehly a injekční stříkačky do kontejneru na likvidaci FDA. Vidět Obrázek k . Ne throw away (dispose of) loose needles a syringes in your household tvyrážka.
- Ne try to touch the needle.
- Pokud nemáte nádobu na likvidaci Sharps Clored FDA, můžete použít kontejner pro domácnost, která je:
- vyrobeno z těžkoprávného plastu
- Lze uzavřít s pevně přiléhajícím víkem odolným proti vpichu, aniž by ostřely mohly vyjít
- Během používání vzpřímeně a stabilní
- odolný vůči únikům a
- Správně označené tak, aby varovala před nebezpečným odpadem uvnitř nádoby.
- Pokud je vaše kontejner pro likvidaci SHARPS téměř plný, budete muset dodržovat své pokyny pro komunitu pro správný způsob, jak zlikvidovat svůj kontejner likvidace Sharps. Mohou existovat státní nebo místní zákony o tom, jak byste měli zahodit použité jehly a stříkačky. Další informace o bezpečném likvidaci Sharps a pro konkrétní informace o likvidaci Sharps ve státě, že žijete na webových stránkách FDA na adrese: https://www.fda.gov/safesharpsdisposal.
- Pro bezpečnost a zdraví vás a dalších jehel a používaných stříkaček nesmí být nikdy znovu použity.
- Použité zásobníky a balení s bavlněnými kuličkami z alkoholu mohou být umístěny do odpadků pro domácnost.
- Ne dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Ne recycle your used sharps disposal container.
- Vždy udržujte nádobu Sharps mimo dosah dětí.
Jak mám ukládat Humiru?
- Humira ukládejte do ledničky mezi 36 ° F až 46 ° F (2 ° C až 8 ° C). Uložte Humiru v původním kartonu, dokud není použita k jeho ochraně před světlem.
- Ne Zmrazení Humira. Ne Použijte Humiru, pokud je zamrzlá, i když byla rozmrazena.
- Chlazená humira může být použita až do data exspirace vytištěné na podnosu HUMIRA Karton nebo předplněná stříkačka. Ne Použijte Humiru po datu vypršení platnosti.
- V případě potřeby například při cestování můžete také ukládat Humiru při teplotě místnosti až do 77 ° F (25 ° C) až pro až do 14 dny. Uložte Humiru v původním kartonu, dokud není použita k jeho ochraně před světlem.
- Odhoďte Humiru, pokud byla udržována při pokojové teplotě a nebyla používána uvnitř 14 dny.
- Zaznamenejte datum, kdy nejprve odstraníte Humiru z chladničky v prostorech uvedených na kartonu a dávce.
- Neukládejte humiru v extrémním teplu nebo chladu.
- Ne use a prefilled syringe if the liquid is cloudy discoločervený or has flakes or particles in it.
- Ne drop or crush Humira. The prefilled syringe is glass.
- Udržujte injekční zásoby HUMIRA a všechny ostatní léky mimo dosah dětí.
Pokyny pro použití
Humira ®
(Hu-Mare-ah)
(Adalimumab)
80 mg/0,8 ml 40 mg/0,4 ml 20 mg/0,2 ml a 10 mg/0,1 ml
Jednorázová předběžná stříkačka
Před injekcí: Váš poskytovatel zdravotní péče by vám měl ukázat, jak používat Humiru, než ji poprvé použijete. Zavolejte svému poskytovateli zdravotní péče nebo 1-800-4HUMIRA (1-800-448-6472) Pokud potřebujete pomoc.
Obrázek a: Humira Single-Dělatse Prefille
 |
Důležité informace, které potřebujete vědět před injekcí Humira
Ne Použijte předběžnou stříkačku a zavolejte svého poskytovatele zdravotní péče nebo lékárníka, pokud:
- Kapalina je zakalena zbarvená nebo má vločky nebo částice
- Uběhlo datum vypršení platnosti
- Kapalina byla zamrzlá (i když se rozmrazovala) nebo ponechána na přímém slunečním světle
- Prefikovaná stříkačka byla upuštěna nebo rozdrcena
Udržujte kryt jehly až těsně před injekcí.
Jak mám ukládat Humiru?
- Humira ukládejte do ledničky mezi 36 ° F až 46 ° F (2 ° C až 8 ° C).
- Uložte Humiru v původním kartonu, dokud není použita k jeho ochraně před světlem.
- Ne freeze
- Chlazená humira může být použita až do data exspirace vytištěné na podnosu HUMIRA Karton nebo předplněná stříkačka.
- V případě potřeby například při cestování můžete také ukládat Humiru při teplotě místnosti až do 77 ° F (25 ° C) až pro až do 14 dny.
- Vyhoďte Humiru, pokud byla udržována při pokojové teplotě a nepoužila se uvnitř 14 dny.
- Zaznamenejte datum, kdy nejprve odstraníte Humiru z chladničky v prostorech uvedených na kartonu a dávce.
- Neukládejte humiru v extrémním teplu nebo chladu.
Udržujte injekční zásoby HUMIRA a všechny ostatní léky mimo dosah dětí.
Přečtěte si pokyny na všech stránkách před použitím předběžné stříkačky HUMIRA
Vzít Humira out of the refrigerator.
Dovolená Humira at room temperature for 15 až 30 minut před injekcí.
- Ne Odstraňte kryt jehly a zároveň umožňují Humiru dosáhnout teploty místnosti
- Ne teplé humira jiným způsobem. Například ne zahřejte ji v mikrovlnné troubě nebo v horké vodě.
- Ne Pokud byla kapalina zmrazena (i když se rozmrazovala, použijte předplněnou stříkačku)
 |
Kontrola Datum vypršení platnosti na předběžné etiketě stříkačky. Ne Pokud uplynulo datum vypršení platnosti, použijte předplacenou stříkačku.
Místo následující na čistém rovném povrchu:
- 1 Sjednocná injekční stříkačka a alkohol
- 1 bavlněná koule nebo podložka gázy (není součástí)
- Kontejner likvidace odolných proti propíchnutí (není součástí). Viz krok 9 na konci těchto pokynů pro použití pokynů, jak zahodit (zlikvidovat) vaši předplnitou stříkačku
Umyjte a osušte vaše ruce.
 |
Vybrat místo injekce:
- Na přední straně stehen nebo
- Vaše břicho (břicho) nejméně 2 palce od pupku (břišní tlačítko)
- Odlišné od vašeho posledního webu injekce
Otřete Místo injekce v kruhovém pohybu s tamponem alkoholu.
- Ne vstříkněte oblečením
- Ne Injekční do kůže, která je bolestivá pohmožděná červená tvrdá zjizvení má strie nebo oblasti s plaky psoriázy
 |
Držet Preffikovaná stříkačka v jedné ruce.
Jemně tahat Jehlu jezdí přímo s druhou rukou.
- Vyhoďte kryt jehly pryč
- Ne Dotkněte se jehly prsty nebo nechte jehlu dotknout se čehokoli
 |
Držet Předplněná stříkačka s jehlou směřující nahoru.
- Držet Preffikovaná stříkačka na úrovni očí jednou rukou, takže můžete vidět vzduch v předběžné stříkačce
- Pomocí druhé ruky pomalu tlačit Pluntár v tlaku vzduchu jehlou.
- Na konci jehly můžete vidět kapku kapaliny. To je normální.
 |
Držet Tělo předplněné stříkačky v jedné ruce mezi palcem a indexovými prsty. Držte předběžnou stříkačku v ruce jako tužka.
Ne kdykoli se zatáhněte na píst.
 |
Jemně stisknout Oblast vyčištěné pokožky na místě injekce druhou rukou. Držte pokožku pevně.
Vložit Jehla do kůže v úhlu asi 45 stupňů pomocí rychlého pohybu podobného šipku.
- Poté, co je jehla v pust kůži.
Pomalu tlačit Plunžru po celou dobu, dokud se vstříkne veškerá kapalina a předplněná stříkačka je prázdná.
 |
Když je injekce dokončena, pomalu vytáhněte jehlu z kůže a přitom udržujte předplnitou stříkačku ve stejném úhlu.
Po dokončení vstřikování umístěte bavlněnou kuličku nebo gázovou podložku na kůži místa vstřikování.
- Ne třít
- Mírné krvácení v místě injekce je normální
 |
Jak bych měl nakládat s použitými předplněnou injekční stříkačkou Humira?
- Okamžitě po použití vložte použité stříkačky a ostřely jehly do nádoby na likvidaci FDA. Ne throw away (dispose of) loose needles a syringes in the household tvyrážka.
- Pokud nemáte nádobu na likvidaci Sharps Clored FDA, můžete použít kontejner pro domácnost, která je:
- vyrobeno z těžkoprávného plastu
- Lze uzavřít s pevně přiléhajícím víkem odolným proti vpichu, aniž by ostřely mohly vyjít
- Během používání vzpřímeně a stabilní
- odolný vůči únikům a
- Správně označené tak, aby varovala před nebezpečným odpadem uvnitř nádoby.
- Pokud je vaše kontejner pro likvidaci SHARPS téměř plný, budete muset dodržovat své pokyny pro komunitu pro správný způsob, jak zlikvidovat svůj kontejner likvidace Sharps. Mohou existovat státní nebo místní zákony o tom, jak byste měli zahodit použité jehly a stříkačky. Další informace o bezpečné likvidaci Sharps a konkrétních informacích o likvidaci Sharps ve státě, ve kterém žijete na webových stránkách FDA na adrese: https://www.fda.gov/safesharpsdisposal.
- Ne dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Ne recycle your used sharps disposal container.
 |
Do domácího odpadku může být umístěna bavlněná koule z alkoholu a balíčku a balení jehly.
Otázky týkající se používání jednorázové stříkačky HUMIRA
Co když jsem neobdržel osobní školení od poskytovatele zdravotní péče?
- Zavolejte svému poskytovateli zdravotní péče nebo 1-800-4HUMIRA (1-800-448-6472) nebo navštivte www.humira.com, pokud potřebujete pomoc
Co když nemám FDA vyčištěný nádoba na likvidaci Sharps nebo správný kontejner pro domácnost?
- Volání 1-800-4HUMIRA (1-800-448-6472) pro bezplatný kontejner s ostře na FDA
Vždy Udržujte předběžnou stříkačku a nádobu na likvidaci Sharps mimo dosah dětí.
Uchovávejte záznam dat a umístění vašich injekcí. Chcete -li si pamatovat, kdy si vzít HUMIRA označit svůj kalendář předem.
 |
 |
Tyto pokyny pro použití byly schváleny americkou správou potravin a léčiv.